Tuberculosis, primero tratar la respuesta inflamatoria
La tuberculosis sigue siendo una causa importante de discapacidad y muerte en el mundo, con un estimado de 8,6 millones de personas enfermas y 1,3 millones muertas a causa de la enfermedad en 2012, según la Organización Mundial de la Salud. Aunque es curable, el cumplimiento del tratamiento es difícil, ya que requiere tomar medicamentos antibióticos durante al menos seis meses y, a veces hasta dos años. La mala adherencia a la medicación y otros factores han dado lugar a cepas resistentes, y en la actualidad no existe ninguna vacuna eficaz contra la afección.
La respuesta inflamatoria de la tuberculosis
Se ha escrito ampliamente sobre la tuberculosis, desde la época de Hipócrates y se mantiene muy presente entre nosotros en el siglo 21. El patógeno infecta aproximadamente a un tercio de la población mundial y probablemente mate a más de 1,3 millones de personas anualmente. El tratamiento más simple tarda unos 6 meses. Como en muchas otras enfermedades infecciosas, los regímenes de terapia se complican cada vez más por la aparición de patógenos resistentes a los fármacos. Son pocas las nuevas terapias antimicobacterianas, y existe una real perspectiva de propagación de enfermedades intratables. Por lo tanto, diferentes aproximaciones a un tratamiento más efectivo amerita un examen minucioso y dettallado.
Muchos especialistas se enfrentan a la dificultad de crecimiento de los cultivos de muestras humanas, incluso aquellos obtenidos a partir de pacientes con extensos síntomas clínicos. Por lo tanto, parece que un número relativamente pequeño de organismos de Mycobacterium tuberculosis puede iniciar una cascada inflamatoria que genera gran daño en los tejidos, complicaciones y muerte. Esto es particularmente cierto en la enfermedad extrapulmonar y en niños. La respuesta inflamatoria innata al complejo M. tuberculosis es multifacético e involucra la producción de muchas células inflamatorias, como monocitos y neutrófilos, y diversos mediadores, como las citocinas y prostaglandinas. Esta respuesta a la infección conduce en última instancia a cambios patológicos que causan los síntomas.
Katrin D. Mayer-Barber y colegas (Nature 2014; 511:99-103) recientemente observaron un aumento de la carga bacteriana de M. tuberculosis y de niveles elevados de eicosanoides (que incluyen la prostaglandina E2 [PGE2] y los leucotrienos) en ratones que son deficientes en interleucina-1, citoquina proinflamatoria clave que está sobre-regulada tempranamente en la respuesta inmune innata humana en la tuberculosis. Entre sus muchos efectos, conduce a la producción de PGE2 en una ruta que requiere la enzima ciclooxigenasa-2 (COX-2). Al utilizar inhibidores de la COX-2, los investigadores fueron capaces de bloquear el control de la infección por micobacterias en macrófagos y luego restaurarla con PGE2.
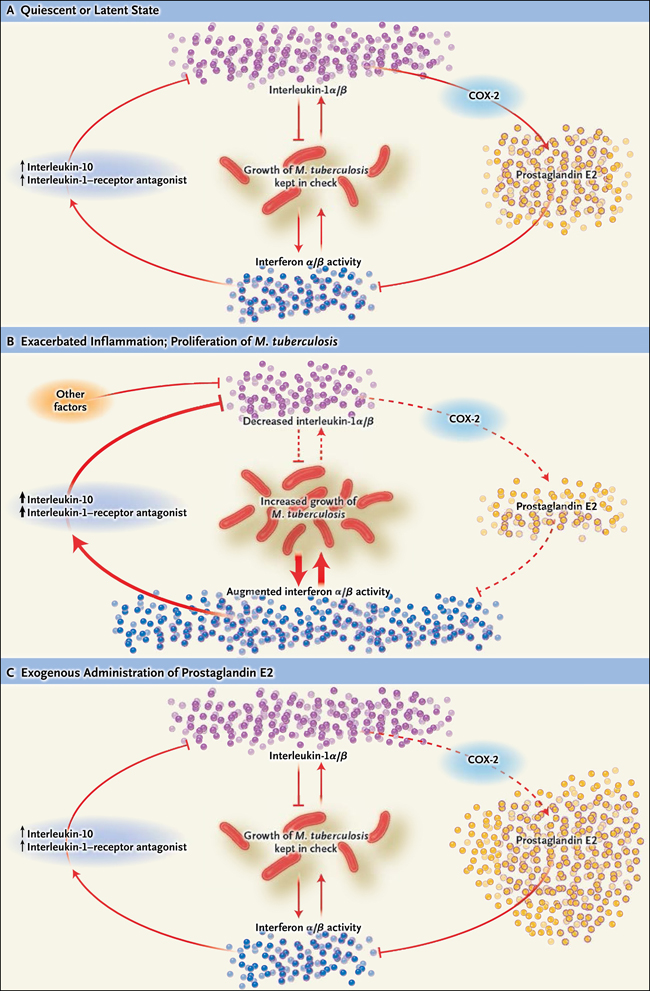
Posteriormente, investigaron el papel de los interferones tipo I sobre la interleucina-1. Desde hace poco, se ha podido conocer la función de dos interferones tipo I (interferón-α y el interferón-β) en el bloqueo de una serie de mecanismos de defensa del huésped contra la tuberculosis. Estos interferones pueden inhibir la señalización de la interleuquina-1 mediante la inducción de los mediadores inmunosupresores de la interleucina-10 y del antagonista de los receptores de la interleucina-1. De hecho, demostraron que la interleuquina-1 y los interferones tipo I se oponen entre sí a través de vías dependientes de prostaglandina (figura 1A). Por ejemplo, al neutralizar los interferones tipo I se reduce el crecimiento de micobacterias en los macrófagos deficientes en el receptor de interleuquina-1. Y los ratones con deficiencia de receptores de la interleucina-1 y de interferón-α son menos susceptibles a M. tuberculosis que los deficientes sólo para el receptor de la interleucina-1.

Figura 1: terapia dirigida al huésped en la tuberculosis
En el estado de reposo o latente, hay un equilibrio entre la interleucina-1 y los interferones tipo I, en parte mediado por la prostaglandina E2, impidiendo la inflamación incontrolada. Cuando se mantiene este equilibrio, el crecimiento de Mycobacterium tuberculosis se mantiene bajo control (panel A). Si éste se altera por factores que reprimen la secreción de interleucina-1, como puede ocurrir en pacientes tuberculosos, la actividad sin oposición de dos interferones tipo I (interferón-α y el interferón-β) puede sobrevenir (panel B). Esto suprime aún más la secreción de interleuquina-1α y de interleucina-1β en un proceso regulado por la actividad de la interleucina-10 y del antagonista del receptor de la interleucina-1-. Esto, a su vez, bloquea la producción de PGE2 y de la enzima ciclooxigenasa-2 (COX-2). El ciclo inflamatorio que es impulsado por el interferón-α y el interferón-β (indicado por el aumento del grosor de la flecha), lleva a un mayor crecimiento de M. tuberculosis. Otras vías inflamatorias innatas probablemente son controladas por estos mediadores. También se muestra la consecuencia de la terapia dirigida al hospedador con PGE2, que restaura el ciclo al estado no inflamatorio (panel C).
En este punto, los investigadores realizaron dos pasos importantes. En primer lugar, observaron dos poblaciones de pacientes con tuberculosis, una en India y otra en China. Encontraron que los individuos con enfermedad avanzada (medido por el crecimiento de M. tuberculosis a partir de muestras de esputo) y aumento de la participación de la condición (caracterizado por cambios bilaterales versus unilaterales en la radiografía de tórax) tenían niveles elevados de interferones tipo I y eicosanoides, bajos niveles de interleucina 1. Estos hallazgos contrastan con concentraciones relativamente bajas de la proteína C reactiva (altos niveles pueden reflejar una enfermedad más grave) en los pacientes con enfermedad avanzada y el hecho de que las mediciones se realizaron en muestras de plasma, en lugar de muestras de esputo inducido o lavado broncoalveolar en el sitio de la infección pulmonar. En segundo lugar, propusieron y probaron lo que ellos llamaban la terapia dirigida que apuntaba a la vía inflamatoria (fig. 1B). Entonces, se trataron ratones infectados que carecían de los genes que codifican la interleuquina-1α y la interleucina-1β con PGE2 y zileuton, un inhibidor de la enzima 5-lipoxigenasa (catalizador de un paso clave en la síntesis de leucotrienos). Tal tratamiento resultó en una reducción de la carga bacteriana y de daño pulmonar, y el aumento de la supervivencia de los animales. Las terapias basadas en PGE2 también fueron eficaces cuando los interferones tipo I fueron reactivaros.
¿Este estudio apunta a un nuevo enfoque para el tratamiento de la infección tuberculosa? Es posible, pero todavía se necesita saber mucho más, por ejemplo, si los interferones tipo I tienen un efecto patogénico importante en la infección humana. Como señalan los autores, puede haber otras vías dependientes de la interleucina-1, e independientes de PGE2 que podrían estar involucradas en la respuesta del huésped. Además, las características patológicas inmunes de la tuberculosis en ratones son muy diferentes a las de los humanos, ya que la enfermedad murino no se caracteriza por la destrucción del tejido pulmonar que conduce a la cavitación, que es el hallazgo clásico en tales pacientes. Es probable que se necesite un nuevo tratamiento para influir en las vías que activan enzimas tales como las metaloproteinasas de la matriz, que son los probables efectores del daño tisular en la tuberculosis. Estos resultados también podrían explicar en parte el limitado papel de los fármacos antituberculosos de segunda línea en la curación de los infectados, ya que hay cierta evidencia que inhiben la PGE2.
Sin embargo, en su aspecto más notable, el estudio del grupo de Katrin D. Mayer-Barber demuestra en un modelo animal que las terapias dirigidas contra la respuesta inflamatoria innata de acogida pueden alterar el resultado de la tuberculosis - un enfoque que difiere fuertemente de los tratamientos farmacológicos comunes dirigidos a los patógenos.
Fuente bibliográfica
Targeting the Inflammatory Response in Tuberculosis
Jon S. Friedland, Ph.D., F.R.C.P.
Department of Infectious Diseases and Immunity, Imperial College London, London.
DOI: 10.1056/NEJMcibr1408663