Tratando la sobrecarga férrica
El exceso de hierro se produce cuando, después de muchos años, el cuerpo absorbe una gran cantidad y éste se acumula en los tejidos de órganos tales como el corazón y/o hígado. Es una enfermedad crónica grave que debe ser diagnosticada y tratada adecuadamente, ya que puede llevar a la hemocromatosis, la cual es potencialmente mortal. Mientras que la mayoría de los casos de hemocromatosis son de origen genético, existen otras causas no genéticas que pueden ser culpables. Estas incluyen complicaciones de otros trastornos de la sangre, la terapia transfusional crónica, hepatitis crónica y la ingesta excesiva de hierro.
El tratamiento preferido para la reducción de los niveles de hierro en pacientes con hemocromatosis se llama flebotomía terapéutica. Es simplemente la eliminación de la sangre del cuerpo y cuando se aplica de forma temprana, evita gran parte del daño que pudiese provocar el exceso férrico. El curso habitual del tratamiento consiste en la eliminación de una unidad de sangre entera, una vez o dos veces por semana, hasta que se elimine todo el exceso. Los niveles de hierro en la sangre son controlados continuamente durante todo el tratamiento, y la duración y frecuencia se determina por la edad del paciente, género, motivo de diagnóstico y severidad de los síntomas. Una vez que se alcanzan los niveles normales, la frecuencia de la flebotomía ser reduce a tres o cuatro veces al año dependiendo de la sintomatología y los niveles de hemoglobina y ferritina en suero.
Cómo controlar la hemocromatosis
La acumulación excesiva de hierro puede causar enfermedad y muerte en los pacientes con hemocromatosis hereditaria y β-talasemia. Hay dos opciones para tratarla: la flebotomía de la hemocromatosis y el uso de quelantes de hierro para β-talasemia, anemia que coexiste con el exceso de hierro. Dos artículos recientes proporcionan pruebas para considerar un nuevo enfoque en relación al desarrollo de tratamientos dirigidos a estas condiciones.
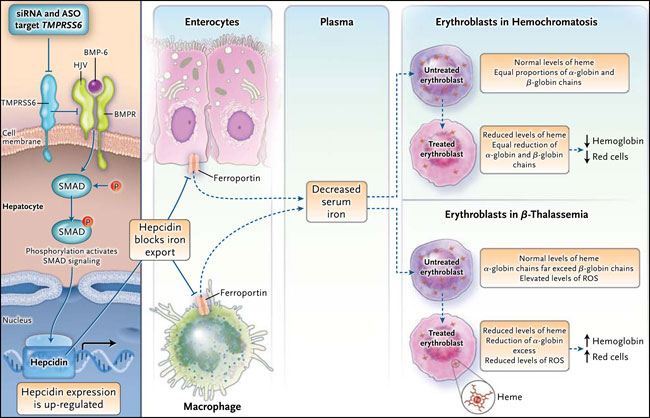
La deficiencia del péptido hepcidina, el regulador central de la homeostasis férrica, es el acontecimiento molecular clave que provoca su acumulación. La hepcidina restringe la expresión en la superficie de la ferroportina exportadora de hierro en los macrófagos y enterocitos - células que liberan hierro al plasma. La hepcidina es sobre-regulada por una vía de señalización que a su vez está inhibida por la proteasa transmembrana TMPRSS6 en los hepatocitos (figura 1). En la hemocromatosis, la vía de activación de la hepcidina es defectuosa, y la severidad de la sobrecarga de hierro se correlaciona con el grado de deficiencia de hepcidina. En homocigotos para la β-talasemia, un trastorno genético heredado, la tasa de absorción de hierro es elevada y la sobrecarga se desarrolla con el tiempo, por lo general debido a transfusiones de sangre. El exceso de hierro puede ocurrir en la β-talasemia con independencia de las transfusiones sanguíneas, ya que la hepcidina es suprimida por señales (aún desconocidas) estimuladas por una eritropoyesis anormalmente amplificada e ineficaz.
La vía de activación de la hepcidina representa hoy un objetivo para los tratamientos dirigidos a modular las concentraciones de hierro. Los miméticos de la hepcidina (mini hepcidinas) se han mostrado prometedores en la reducción de la acumulación de hierro en animales con deficiencia de hepcidina. Los estudios genéticos en los que Tmprss6, que codifica un inhibidor de la hepcidina, se suprimía en modelos de ratón para la hemocromatosis (HFE -/-) y β-talasemia (Hbb th3/+) demostraron que puede prevenirse la sobrecarga de hierro. Inesperadamente, la anemia era más suave, la eritropoyesis menos aberrante, y la esplenomegalia se reducía en ratones con β-talasemia y ablación de Tmprss6, en comparación con los animales con β-talasemia y Tmprss6 intactos.

Figura 1: inhibición de TMPRSS6
La inhibición de Tmprss6 aumenta la señalización mediada por SMAD dentro del hepatocito (activada por la fosforilación [P]) y sobre-regula la expresión de la hepcidina en los hepatocitos. La hepcidina secretada bloquea la salida de hierro que son absorbidos por enterocitos y reciclados por macrófagos mediante la degradación de la ferroportina. En el plasma, disminuye el hierro y es liberado a todos los órganos, incluyendo el hígado (no se muestra). El efecto de los bajos niveles de hierro en la eritropoyesis es diferente en la hemocromatosis y en la β-talasemia. La imagen muestra los resultados finales en la hemoglobina y glóbulos rojos. ASO: oligonucleótido alelo-específico, BMP-6: proteína morfogenética de hueso 6, BMPR receptor de la proteína morfogenética ósea, HJV: hemojuvelina, ROS: especies reactivas del oxígeno y siRNA: pequeños ARN de interferencia.
Paul J. Schmidt y colaboradores (Blood 2013; 121:1200-1208) suprimieron la expresión de Tmprss6 mediante la inyección de ARN de silenciamiento formulado en nanopartículas lipídicas en ratones HFE -/- y ratones Hbb thal3/+. Por otro lado, Shulin Guo y colegas (J Clin Invest 2013; 123:1531-1541) también se centraron en Tmprss6, en ambos modelos de ratón pero utilizaron oligonucleotidos antisentido para inhibir la expresión de Tmprss6. Ambos grupos de investigadores observaron una eficiente y persistente baja en la regulación de Tmprss6 y una elevación en los niveles de hepcidina. En los animales con hemocromatosis se determinó una caída de los niveles de hierro sérico y hepático, y una leve anemia por deficiencia férrica relacionada con la dosis fue el único efecto secundario observado (figura 1). Este hallazgo no es sorprendente, teniendo en cuenta que la pérdida de función de Tmprss6 en seres humanos genera concentraciones elevadas de hepcidina y anemia por falta de hierro.
Con la inyección de cualquiera de los agentes anti-Tmprss6 en ratones Hbb th3/+, el hierro corporal total se redujo y se distribuyó desde la células del parénquima a los macrófagos. Los autores observaron una mejora de la anemia en estos animales, consistente con el hallazgo en ratones talasémicos deficientes para Tmprss6. El nivel de hemoglobina aumentó aproximadamente en 2 g por decilitro, la morfología eritrocitaria mejoró y la esplenomegalia se redujo. ¿Cómo pudo pasar esto?
En la β-talasemia, la síntesis coordinada de hemo y α-globina y de las β-cadenas de globina es errónea, lo que lleva a un exceso de cadenas insolubles de α-globina que dañan gravemente a los eritroblastos en vías de maduración a través de la formación de especies reactivas de oxígeno. El tratamiento de los animales talasémicos con agentes anti-Tmprss6 conduce a una disminución de las cadenas alfa asociadas a membrana, una disminución de la α-globina y agregados tóxicos hemo, y a una disminución en la formación de especies reactivas de oxígeno (figura 1), lo que aumenta la maduración y la supervivencia de los glóbulos rojos. Este efecto en las células eritroides talasémicas no se genera directamente por el silenciamiento de Tmprss6; más bien, es el resultado de los bajos niveles de hierro circulantes causados por el aumento de la producción de hepcidina (figura 1). Se está acumulando evidencia que restricción de hierro beneficia paradójicamente la eritropoyesis talasémica, probablemente debido a que una menor velocidad en la formación de hemo se asocia con una tasa más lenta de traducción del ARN mensajero que codifica la α-globina. La quelación del hierro en pacientes con talasemia no dependiente de transfusión no mejorar la anemia, sin embargo, los actuales protocolos de quelación de hierro no logran su control.
Las terapias orientadas a la hepcidina parecen prometedoras, con efectos secundarios poco notables a corto plazo. ¿Cómo podrán ser trasladadas a la práctica clínica? La inhibición de Tmprss6 para el control de la sobrecarga de hierro está dirigida al mecanismo de la enfermedad de una manera que es más fisiológica que la flebotomía. Sin embargo, a diferencia de ésta y la quelación del hierro, el aumento de la hepcidina no elimina o excreta el hierro. Por lo tanto, no se podrían sustituir los tratamientos tradicionales en pacientes con hemocromatosis severa y talasemia dependiente de transfusiones. Más bien, se podría usar en combinación o como manejo alternativo en pacientes seleccionados con hemocromatosis para quienes la flebotomía no es opción de tratamiento. Los mejores candidatos para las nuevas terapias pueden ser pacientes con talasemia no-dependientes de transfusiones y aquellos con talasemia que han sido aloinmunizados y no pueden someterse a una transfusión segura. El riesgo de deficiencia de hierro como consecuencia de los nuevos tratamientos debe ser evaluado cuidadosamente. El desarrollo de biomarcadores que discriminen la eritropoyesis por restricción de hierro vesrus la deficiencia de podría ayudar en el seguimiento de estas terapias.
Fuente bibliográfica
Treating Iron Overload
Clara Camaschella, M.D.
Vita-Salute University and San Raffaele Scientific Institute, Milan.
DOI: 10.1056/NEJMcibr1304338