Reprogramación terapéutica para la piel de cristal
La epidermolisis bullosa es un grupo de trastornos hereditarios caracterizados por la formación de ampollas en respuesta a un trauma mecánico. Históricamente, los subtipos se han clasificado de acuerdo a la morfología de la piel. Los recientes descubrimientos de las bases moleculares de la epidermólisis ampollosa se han traducido en el desarrollo de nuevas herramientas de diagnóstico, incluyendo el prenatal y las pruebas de preimplantación. Sobre la base de una mejor comprensión de la zona de la membrana basal y los genes responsables, los nuevos tratamientos (por ejemplo, terapia génica o de proteínas) pueden proporcionar soluciones a la fragilidad de la piel de los pacientes que padecen la condición.
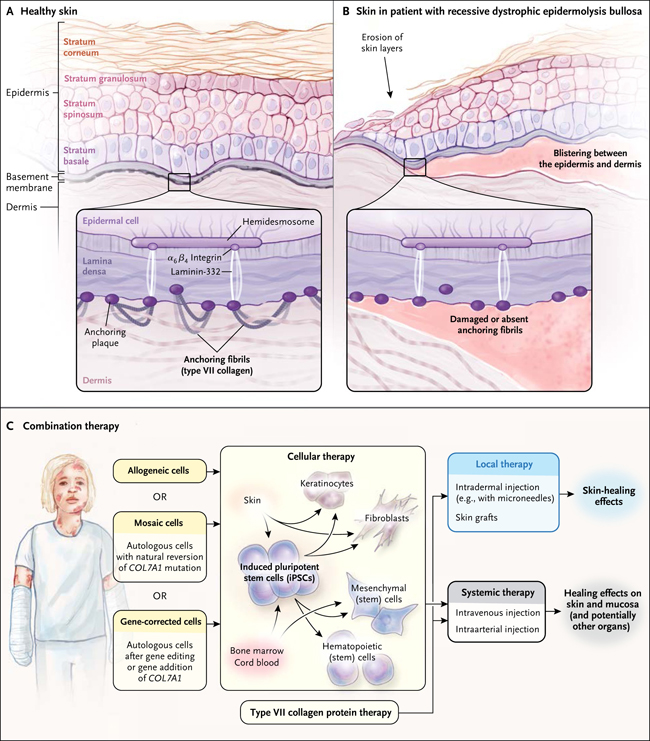
Terapia combinada
La mayoría de nosotros mantenemos una piel sana. Es nuestra frontera física con el mundo externo, que además, es duradera y se auto-repara. Rica en terminaciones nerviosas, proporciona información constante acerca del medio ambiente, manteniendo la humedad y defensa contra gérmenes nocivos. Sin embargo, en los pacientes con epidermólisis bullosa, un trastorno que provoca fragilidad de la piel, las membranas mucocutáneas se dañan severamente en respuesta a los esfuerzos mecánicos normales de la vida diaria.
En la piel normal, tres moléculas de colágeno polimerizado tipo VII (C7), interactúan con las proteínas dérmicas y epidérmicas, y forman el componente principal de las fibrillas de anclaje. Como un “velcro” biológico, las fibrillas conectan las capas de la piel mediante la ampliación de la membrana basal cutánea y se enganchan en las fibras de colágeno intersticial de la dermis papilar. En la epidermolisis bullosa distrófica recesiva (RDEB, por sus siglas en inglés), mutaciones bialélicas hacen perder la función del gen que codifica a C7 (COL7A1) resultado severamente disminuido o ausente. Sin fibrillas de anclaje en la unión dermo-epidérmica, los pacientes con RDEB tienen ampollas graves en la piel, alopecia, distrofia ungueal, erosiones corneales y heridas en la mucosa. Este daño es extremadamente doloroso, con gran sensibilidad a la presión, al calor y al frío, pérdida de líquido y aumento de infección local y sistémica. El compromiso de la parte superior del esófago conduce a la estenosis, pérdida crónica de sangre y a un deficiente desarrollo. La frecuente lesión de la piel y la aberrante reparación del tejido causan mala cicatrización y deformidades en dedos de manos y pies. Las persistentes heridas que nunca sanan dejan a los niños, algunos de tan sólo 6 años de edad, con una gran tendencia a agresivos carcinomas de células escamosas.
El sufrimiento extremo y los diversos efectos de este trastorno han motivado a los equipos de investigación a determinar terapias mediante muchos enfoques, por ejemplo, con técnicas génicas, de proteínas o basadas en células. El hecho de no obtener una cura total ha impulsado mayores esfuerzos hacia la búsqueda de un tratamiento adecuado.
Uno de estas iniciativas ha sido recientemente descrita por Vittorio Sebastiano y colaboradores (Sci Transl Med 2014; 6(264):264ra163). En primer lugar, ellos crearon células madre pluripotentes inducidas a partir de fibroblastos y queratinocitos; a continuación, corrigieron genéticamente la mutación COL7A1 en estas células, utilizando un virus altamente recombinogénico para mediar la reparación. En segundo lugar, se utilizó de forma combinada la secuenciación del genoma y la bioinformática para caracterizar las alteraciones genómicas en las células madre de RDEB, incluyendo cambios en genes que están involucrados en la patogénesis del carcinoma de células escamosas. Esto permitió seleccionar células corregidas y minimizar los potenciales riesgos de la terapia génica. En tercer lugar, se derivaron láminas de queratinocitos a partir de las células madre pluripotentes inducidas, las que posteriormente se injertaron, demostrándose de manera concluyente que C7 se expresaba y era funcional, tanto in vitro como in vivo. En resumen, el equipo de Vittorio Sebastiano ha podido establecer un nuevo y refinado proceso para la generación de injertos autólogos de piel en el tratamiento de las heridas en la RDEB, comenzado a establecer normas para la fabricación de células en relación a futuros ensayos clínicos.

Figura 1: terapia combinada para la epidermolisis bullosa
Diferentes enfoques terapéuticos son posibles para la epidermólisis bullosa, aunque ninguno ha tenido éxito completo. Las terapias locales incluyen la inyección intradérmica, uso de microagujas e injertos de piel, como el propuesto en este estudio. Tales métodos han demostrado ser eficaces en la curación de heridas superficiales, pero no cubren todas las manifestaciones físicas de la enfermedad. La curación más probable pasa por aplicar un enfoque combinado de las terapias locales y sistémicas, de forma individualizada para cada paciente.
Aunque este avance sustantivo da esperanza de que las dolorosas y molestas lesiones de piel se puedan curar, no resuelve otras manifestaciones clínicas, porque las heridas ocurren en cualquier parte de la superficie y en cualquier momento, y el injerto en todas las zonas no es posible. Asimismo, los biofilms para las heridas son reto importante para el éxito de los injertos. Además, la RDEB es una patología sistémica que a menudo conduce a una lesión de la mucosa oral y en la parte superior del esófago, córnea y el riñón, donde el injerto no puede llegar. Por último, el riesgo de carcinoma de células escamosas, típicamente agresivo, metastásico y letal, puede persistir mientras las heridas se repiten con frecuencia.
Por lo tanto, es necesario un enfoque sistémico, que sea capaz de modificar el curso de la RDEB utilizando poblaciones de células hematopoyéticas que puedan circular por todos los tejidos del cuerpo a lo largo de la vida. La clave es una plataforma más amplia de terapia combinada en estos pacientes, generada por la inteligencia colectiva y el trabajo colaborativo de los investigadores (fig. 1). En este escenario, adiciones o correcciones a COL7A1, sustitución de la proteína C7 y la terapia celular, podrían aplicarse de forma local o sistémica, una vez o en serie, sola o en combinación con otras intervenciones que cooperativamente establezcan un conjunto de opciones que permitan cambios personalizados y clínicamente significativos en niños y adultos con RDEB.
Fuente bibliográfica
A Biologic Velcro Patch
Jakub Tolar, M.D., Ph.D., and John E. Wagner, M.D.
Department of Pediatrics, Division of Blood and Marrow Transplantation, and Stem Cell Institute, University of Minnesota, Minneapolis.
N Engl J Med 2015; 372:382-384