Neuroinflamación y Alzheimer
Con más de 35 millones de personas en todo el mundo afectadas, la enfermedad de Alzheimer es la forma más común de demencia y una amenaza creciente para la salud pública. Es un trastorno primario neurodegenerativo que se reconoce por tres características patológicas principales: la pérdida neuronal, ovillos neurofibrilares y placas compuestas de Aβ1. Sobre la base de la identificación de mutaciones causantes de la enfermedad familiar de inicio temprano, se ha propuesto la hipótesis de la cascada amiloide, que sitúa la acumulación de Aß en el centro de la patogénesis de la enfermedad de Alzheimer.
A partir del análisis de las mutaciones causantes, la investigaciones se han centrado en el desarrollo de modelos de ratones transgénicos con la finalidad de poder estudiar de forma más fidedigna las características patológicas de las placas de la proteína beta-amiloide. Dichos animales son útiles en el estudio de la amiloidosis, así como los mecanismos básicos de la enfermedad in vivo.
Citoquinas neuroinflamatorias y el Alzheimer
La neuroinflamación, definida como una activación microglial con una excesiva expresión de citoquinas inmunes, rápidamente adquiere la condición de ser el "principal culpable" en la conexión sin resolver del elevado riesgo de la enfermedad esporádica con la lesión cerebral traumática, las infecciones sistémicas, el envejecimiento normal y varios trastornos neurológicos. La neuroinflamación también parece ser un contribuyente importante en personas con síndrome de Down (debido a la gran cantidad de genes que participan) y en individuos con mutaciones genéticas que afectan la proteína precursora amiloide (APP) o presenilina.
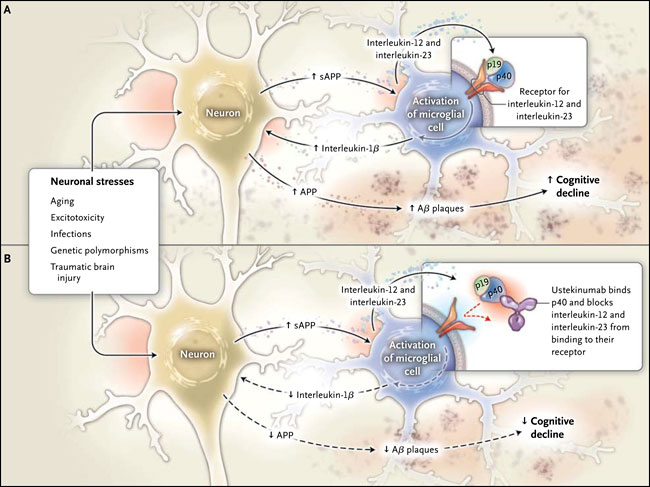
Las moléculas y las vías que median la inflamación asociada con la enfermedad de Alzheimer están siendo fuertemente estudiadas. Un avance en esta área ha sido descrito por Johannes vom Berg y colaboradores (Nat Med 2012; 18:1812-9), quienes utilizaron un modelo de ratón para el Alzheimer con tal de investigar la función de las citoquinas proinflamatorias en la patogénesis de la condición (fig. 1). Los resultados demostraron que la amortiguación de la expresión y señalización de las citocinas interleucina-12 e interleucina-23 estaba asociada con disminuciones de la activación microglial, a nivel de β-amiloide soluble (Aß), y en la cantidad total de placa Aß. Estas conclusiones son consistentes con estudios anteriores que han relacionado la activación microglial con un mayor expresión de la interleucina-1 (que regula la señalización de las interleucinas 12 y 23), además de la expresión de APP (cuando se escinde genera Aß), el desarrollo de placas Aß, y la activación de la microglia en el cerebro de pacientes con Alzheimer.

Fuente bibliográfica
Neuroinflammatory Cytokine Signaling and Alzheimer’s Disease
W. Sue T. Griffin, Ph.D.
Donald W. Reynolds Department of Geriatrics and Institute on Aging, University of Arkansas for Medical Sciences, and the Geriatric Research, Education, and Clinical Center (GRECC) at the Central Arkansas Veterans Healthcare System, Little Rock.
DOI: 10.1056/NEJMcibr1214546