MED12 y el control de la respuesta a fármacos contra el cáncer
A menudo la terapia del cáncer se ve obstaculizada por la rápida aparición de resistencia a los medicamentos. Esto es cierto no sólo para las quimioterapias convencionales, sino también para la nueva generación de fármacos dirigidos a aquellos componentes que están mutados o desregulados en las células tumorales. Por ejemplo, el tratamiento para del cáncer de pulmón de células no pequeñas metastásico (NSCLC) que alberga mutaciones en el gen que codifica el receptor del factor de crecimiento epidérmico (EGFR) da lugar a un aumento significativo de la supervivencia libre de progresión. Sin embargo, estas respuestas son generalmente de corta duración, lo que resulta en menos beneficios para el paciente en términos de supervivencia global.
Se sabe que esta falta de resultados positivos a largo plazo se debe a la aparición de variantes resistentes a los medicamentos.
Resistencia en cánceres causados por oncogenes
La aparición de resistencia a los fármacos se produce comúnmente en los tumores impulsados por oncogenes - por ejemplo, en los cánceres de pulmón con la activación de la mutación de EGFR o con una translocación que fusiona los genes EML4 y ALK, y en melanomas con la activación de mutaciones de BRAF. En pacientes con este tipo de tumores, el desarrollo de resistencia a los medicamentos causa una eventual recaída a pesar de la buena respuesta inicial y larga supervivencia libre de progresión. La terapia estándar en estos individuos, basada en la selección de una mutación controladora para el tratamiento con un fármaco, está limitada por los circuitos de retroalimentación y la diafonía entre las vías de señalización. Estos datos sugieren que una mayor investigación podría identificar las vías más importantes de una posible oncoproteína objetivo lo que conduciría a un mejor resultado al permitir el diseño racional de terapias combinadas.
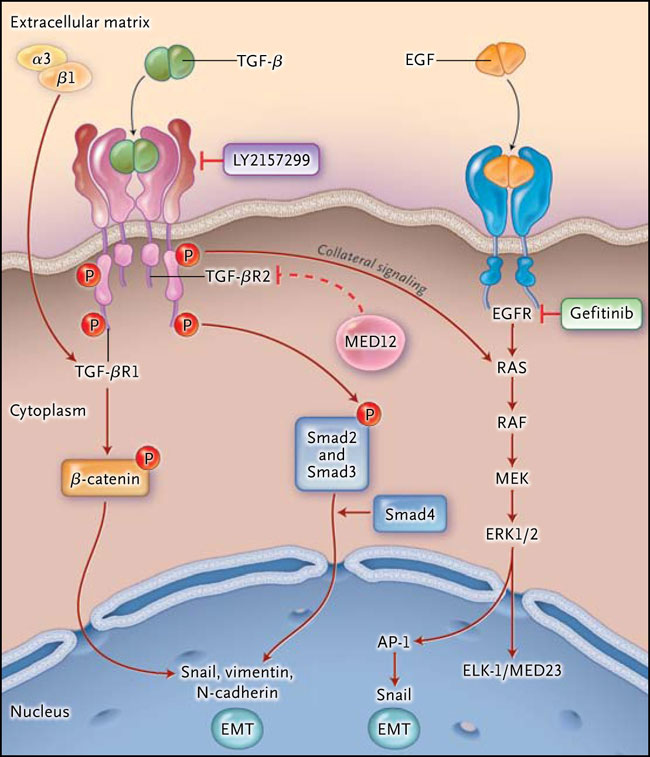
Sidon Huang y colegas (Cell 2012; 151:937-50) han informado recientemente que MED12, un componente del complejo mediador transcripcional, modula la respuesta a los inhibidores de EGFR, ALK, y BRAF mediante la regulación negativa del receptor de factor de crecimiento transformante β 2 (TGF-βR2). La inhibición de la señalización de TGF-βR restaura la capacidad de respuesta de las drogas en células deficientes MED12, sugiriendo un nuevo mecanismo de letalidad en tumores resistentes a fármacos que carecen de MED122 (fig. 1).
El complejo mediador es un regulador transcripcional 26-S que une las secuencias reguladoras de ADN a la ARN polimerasa II del complejo de transcripción-iniciación. A través de interacciones físicas entre los factores de transcripción y las subunidades mediadoras específicas, el mediador funciona como un centro integral para la canalización de diferentes vías de señalización. Por ejemplo, MED23 se desempeña como un objetivo nuclear en la vía RAS-RAF-MEK-ERK y, junto con su pareja de unión la proteína ELK-1, actúa como regulador crítico en esta vía de señalización en tumores con mutaciones en EGFR, RAS o BRAF 3 (fig. 1). MED12 es un transductor crítico de la información normativa transmitida por los factores de transcripción activados por señales vinculadas a diversas vías de desarrollo. MED12 citoplasmático interfiere con la glicosilación de TGF-βR2, bloqueando la expresión de la superficie celular del receptor.

Figura 1: MED12 como mediador de la resistencia a los medicamentos en tumores inducidos por oncogenes
MED12, un componente del complejo mediador transcripcional, regula el factor de crecimiento transformante β (TGF-β) mediante la inhibición de la glicosilación de formas inmaduras del receptor de TGF-β 2 (TGF-βR2), evitando de este modo su expresión en la superficie celular. Este estudio nos muestra la pérdida de MED12 en el desarrollo de la resistencia a terapias dirigidas en líneas cancerígenas con la activación de EGFR o mutaciones BRAF o la reordenación de EML4-ALK. La falta de MED12 (línea de trazos) permite la activación de TGF-β mediante la señalización colateral de ERK1/2. El regulador MED23, que se une a ELK1, también puede contribuir a la resistencia tumoral con mutaciones que afectan a la vía RAS-RAF-MEK-ERK. La proteína activadora 1 (AP-1) está regulada por ERK1/2 para promover la transición epitelio-mesenquimal (EMT). En la vía de TGF-β, TGF-βR2 activa a TGF-βR1, que a su vez fosforila a dos Smads (Smad2 y Smad3) y a la Smad más frecuente (Smad4). Las Smads a continuación, translocan al núcleo y regulan la expresión de los genes diana TGF-β. La señalización de TGF-β induce la EMT a través de la fosforilación (P) de β-catenina, que conduce a la sobre regulación de Snail, vimentina y N-cadherina. Otros factores de crecimiento también pueden inducir la EMT, incluyendo algunas integrinas, tales como α3β1. Los autores encontraron que la adición del inhibidor de TGF-β, LY2157299, a los inhibidores de EGFR suprime la resistencia adquirida por la pérdida de MED12 en células PC9 EGFR mutantes. La inhibición de TGF-β por LY2157299 inactiva a ERK y a las Smads, causando la letalidad cuando se combina con gefitinib, el inhibidor de EGFR.
La clave en el estudio de Sidon Huang es que la pérdida de MED12 activa la señalización colateral a través de la vía RAS-RAF-MEK-ERK, causando resistencia a EGFR, ALK y a los inhibidores de BRAF en tumores impulsados por oncogenes. En consonancia con este hallazgo, está la observación que el inhibidor de TGF-β, LY2157299, cuando se asocia con crizotinib o gefitinib, reprime fuertemente la activación de ERK en células H3122 y PC9, que controlan la reordenación de EML4-ALK y las mutaciones de EGFR, respectivamente. La activación de la señalización de ERK por la supresión de MED12 en células de melanoma A375 (que expresan endógenamente la mutación BRAF V600E) confiere resistencia a vemurafenib (un inhibidor de BRAF) y a selumetinib (un inhibidor de MEK).
Un segundo hallazgo importante es que células cancerosas resistentes a fármacos mostraron propiedades similares a la transición epitelial a mesenquimal (EMT), una notable característica de las células sometidas a proliferación, con un aumento en la expresión de Snail, vimentina, y N-cadherina. El estímulo prototípico para EMT es la señalización de TGF-β, que induce la fosforilación de β-catenina y la posterior activación de varios factores de transcripción. Sin embargo, otros factores de crecimiento y receptores de transmembrana, incluyendo integrinas, también pueden inducir la EMT. Las integrinas son receptores de adhesión asociados a la matriz, y la integrina α3β1 forma complejos con la E-cadherina y TGF-βR1; la activación de la integrina α3β1 se requiere para establecer una cadena de señalización intracelular que hace girar a la célula hacia EMT 4 (fig. 1).
Los resultados obtenidos en este artículo subrayan la necesidad de impulsar estrategias que se dirijan a más de una vía en tumores provocados por un oncogén. Inhibidores de TGF-β, como LY2157299, pueden ser particularmente eficaces cuando se combinan con terapias dirigidas apropiadamente en un subconjunto de cánceres molecularmente definidos. El descubrimiento que la pérdida de MED12 confiere resistencia a los medicamentos también descubre un enlace entre mecanismos independientes de la EMT. Se necesita que la investigación clínica incluya muestreos de biopsias seriadas en tumores provocados por oncogenes para poder confirmar que la pérdida de función de MED12 regula la resistencia tumoral.
Fuente bibliográfica
Mediating Resistance in Oncogene-Driven Cancers
Rafael Rosell, M.D., Ph.D.
Catalan Institute of Oncology, Badalona, and the Breakthrough Cancer Research Unit, Pangaea Biotech, Dexeus University Institute, Barcelona
DOI: 10.1056/NEJMcibr1214549