Diabetes tipo 2: un problema de identidad
La diabetes tipo 2 es un trastorno metabólico que se caracteriza por niveles elevados de glucosa en la sangre. Esta compleja enfermedad afecta a más de 170 millones de individuos en todo el mundo y es causada por una combinación de múltiples factores genéticos y de exposición ambiental. Los primeros intentos para descifrar su genética sólo han logrado éxito limitado, con la identificación de algunos loci con asociación convincente. Sin embargo, con la llegada y aplicación de diferentes técnicas genómicas, se ha avanzado mucho en los últimos años en la comprensión molecular de la afección. Por ejemplo, más de una docena de zonas cromosómicas con susceptibilidad presentan asociaciones consistentes y robustas a través de diferentes poblaciones, ejerciendo el negativo efecto mediante la disfunción pancreática de las células beta. Resolver lo anterior, es un gran y urgente desafío para la medicina actual.
Diabetes tipo 2 por des-diferenciación de células beta
La diabetes tipo 2 es el resultado de la combinación entre la resistencia a la insulina y la disfunción de las células beta pancreáticas productoras de insulina. Se cree que la deficiencia es causada por la disfunción celular y disminución de masa. Tal reducción se ha atribuido a una mayor apoptosis de las células beta y por tanto a una menor proliferación. Por consiguiente, muchos esfuerzos de investigación están dirigidos hacia la restauración funcional mediante la prevención de la muerte celular y la inducción de la replicación.
También se ha observado en la diabetes tipo 2 un aumento de la secreción de glucagón desde las células de los islotes alfa, y su papel central en la desregulación metabólica ha sido recientemente enfatizado. Por otra parte, la hiperglucagonemia resulta de la inhibición disminuida de la paracrina en las células alfa por presencia de insulina secretada a nivel local, aunque también se ha postulado un aumento relativo de la cantidad de células alfa.
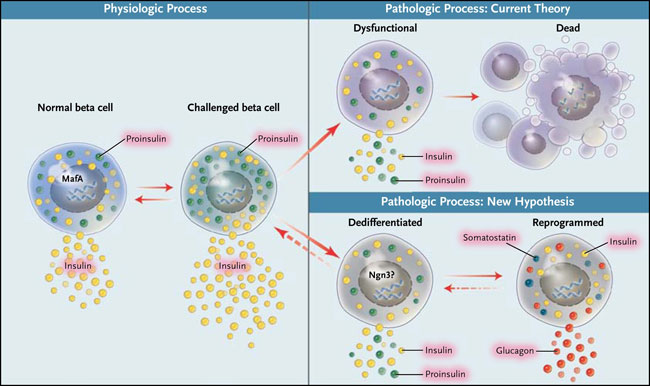
Chutima Talchai y colegas (Cell 2012; 150:1223-34) acaban de postular una posible explicación para la insuficiencia de células beta y la hiperfunción celular alfa en la diabetes 2. Ellos examinaron el efecto del factor de transcripción FoxO1 en las unidades beta, cuya expresión se reduce fuertemente en la hiperglucemia grave. Para estudiar tal fenómeno, los investigadores modificaron genéticamente a ratones para que sus células beta no pudiesen expresar FoxO1. Estos animales eran saludables, sin embargo, el envejecimiento o embarazos múltiples (condiciones que aumentan la presión metabólica en las células beta) causaban hiperglucemia, una reducción dramática de la masa de células beta, y el aumento del número de células alfa. Además, la expresión de la proteína PCSK1 (enzimas convertasa tipo 1) se reducía en las células beta de ratones modificados después de someterlos a estrés, conduciendo a un aumento de proinsulina biológicamente inactiva, lo que también se ha observado en la diabetes tipo 2 no controlada. Por lo tanto, la supresión FoxO1 en células beta ofrece un nuevo modelo para la diabetes inducida por estrés.
También se observó un resultado sorprendente cuando la deleción de FoxO1 se combinó con el permanente marcaje genético de las células beta. Esto demostró que células beta deficientes para FoxO1 no habían muerto sino que habían perdido la expresión de genes clave como los que codifican la insulina, el transportador de glucosa 2, la glucoquinasa y los principales factores de transcripción (fig. 1). Además, las células deficientes de FoxO1 a menudo expresaban hormonas de otros islotes, como el glucagón, pudiendo explicar el aparente aumento de masa en células alfa. Estos resultados sugieren que la aparente pérdida de células positivas para la insulina no reflejaba la simple des-granulación de células beta sometidas a estimulación a largo plazo, sino más bien una pérdida más profunda de la identidad celular.

Fuente bibliográfica
Beta-Cell Dedifferentiation and Type 2 Diabetes
Yuval Dor, Ph.D., and Benjamin Glaser, M.D.
Department of Developmental Biology and Cancer Research, Institute for Medical Research Israel-Canada, Hebrew University–Hadassah Medical School (Y.D.), and Endocrinology and Metabolism Services, Department of Internal Medicine, Hadassah-Hebrew University Medical Center (B.G.) — both in Jerusalem.
DOI: 10.1056/NEJMcibr1214034