Bases de la toxicidad miocárdica por antraciclinas
Si bien no se ha dilucidado completamente el mecanismo subyacente a la citotoxicidad severa por doxorubicina y otras antraciclinas, hay evidencias que los efectos tóxicos del fármaco pueden producirse a través de la formación de radicales libres y el subsiguiente ciclo redox con O2, lo que llevaría a la generación de especies reactivas de oxígeno como el anión superóxido, radicales hidroxilo y el peróxido de hidrógeno. Los tejidos con defensas antioxidantes menos desarrollados, como el corazón, son particularmente susceptibles al daño por radicales de oxígeno inducido por una antraciclina.
Antraciclinas e insuficiencia cardíaca
Desde su descubrimiento hace más de 50 años atrás, las antraciclinas se han convertido en el principal soporte para el tratamiento de muchos tipos de cáncer. Sin embargo, se asocian con insuficiencia cardíaca y exposición acumulativa; en donde la lesión al corazón parece ocurrir con todas las dosis, y biopsias obtenidas horas después de una dosis única de antraciclina (por ejemplo, doxorubicina o daunorubicina) demuestran verdaderos cambios patológicos. Se han invertido muchos esfuerzos en encontrar maneras de prevenir tal cardiotoxicidad, sin embargo, la insuficiencia cardíaca avanzada sigue siendo una consecuencia de la exposición a este tipo de medicamentos. Por otra parte, la insuficiencia sintomática a menudo se produce años después del tratamiento, complicando la evaluación de estrategias preventivas.
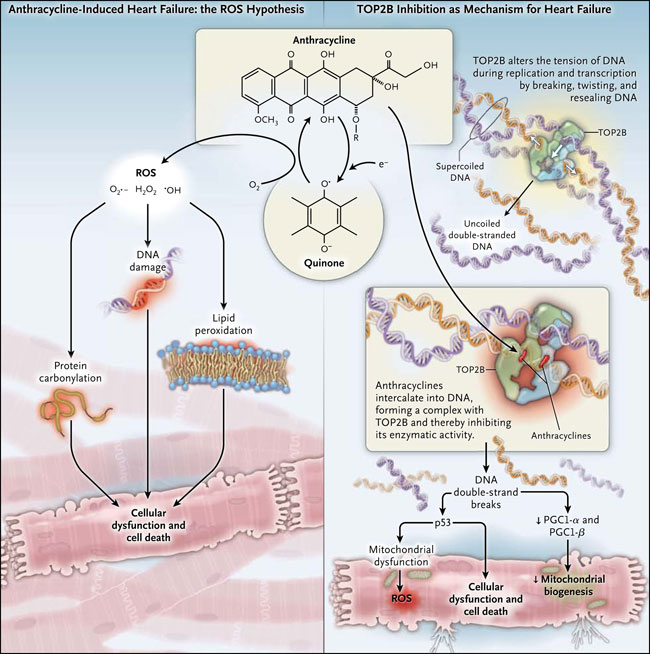
La teoría dominante de cómo las antraciclinas dañan al corazón implica a la generación de especies reactivas de oxígeno, las que dañan al ADN, a proteínas y lípidos, provocando la disfunción celular y la muerte de los miocitos. Sin embargo, los resultados de un nuevo estudio realizado por Sui Zhang y colaboradores (Nat Med 2012; 18:1639-42) sugieren que el primer paso del daño a los miocitos cardíacos es independiente de las especies reactivas de oxígeno y depende de interacciones medicamentosas con un tipo particular de topoisomerasa, una enzima que afecta a la tensión y características topológicas del ADN.
Las antraciclinas interrumpen el crecimiento del tumor mediante la unión y bloqueo de la topoisomerasa II (TOP2). Estas enzimas rompen, tuercen y cierran nuevamente el esqueleto de fosfato del ADN y, de este modo, alteran la tensión de la doble hélice durante la replicación y transcripción. Las antraciclinas se intercalan en el ADN y forman complejos con la TOP2, interrumpen la actividad de la enzima y activan una respuesta al daño del ADN, induciendo la muerte celular. Existen varias formas de topoisomerasas. Las células tumorales se dividen rápidamente y expresan niveles elevados de topoisomerasa II alfa (TOP2A). La TOP2 beta (TOP2B) se expresa por doquier; los cardiomiocitos expresan TOP2B pero no TOP2A. Finalmente, las antraciclinas se dirigen tanto a la TOP2A como a la TOP2B.
Se cree que las antraciclinas dañan a los cardiomiocitos por reacciones que resultan en la formación de radicales libres, que a su vez alterar la función de muchos constituyentes celulares, provocando disfunción y muerte celular. Numerosos estudios en células aisladas y en animales han demostrado efectos cardioprotectores de los antioxidantes, sustentando la hipótesis que, como consecuencia de la exposición a una antraciclina, las especies reactivas de oxígeno afectan negativamente a los cardiomiocitos (fig. 1). Sin embargo, los ensayos clínicos con antioxidantes para la prevención de la lesión cardíaca inducida por antraciclina han sido decepcionantes. No obstante, el dexrazoxano, un compuesto quelante del hierro es capaz de evitar la formación de radicales hidroxilo en presencia de antraciclinas, lo que previene la lesión cardíaca, esto, apoya la teoría respecto a las especies reactivas de oxígeno (el dexrazoxano ha sido aprobado por la Administración de Alimentos y Fármacos de Estados Unidos para la prevención de dicha cardiotoxicidad).

Fuente bibliográfica
Anthracyclines and Heart Failure
Douglas B. Sawyer, M.D., Ph.D.
Vanderbilt University Medical Center, Nashville.
DOI: 10.1056/NEJMcibr1214975