A la caza de la huntingtina mutante
La enfermedad de Huntington tiene la mayor prevalencia de todas las enfermedades neurodegenerativas autosómicas con repetición CAG. El gen de la huntingtina mutante confiere toxicidad atribuida a la proteína mutante, aunque la contribución de su ARNm para el proceso aún no se ha probado completamente. La neuropatología se caracteriza por atrofia neuronal estriatal y cortical. Los pacientes generalmente desarrollan movimientos involuntarios, disfunción cognitiva y cambios de comportamiento en la cuarta década de la vida.
A pesar de la identificación de la huntingtina mutante como la causa genética en 1993, no existe una terapia modificadora de la enfermedad. Varios candidatos terapéuticos con el potencial de alterar la progresión subyacente han sido evaluados en ensayos clínicos. Estos agentes incluyen esquemas neuroprotectores para contrarrestar los efectos tóxicos celulares de la proteína mutante y estrategias celulares de reemplazo para compensar la pérdida de neuronas en el estriado. En el mejor de los casos, tales enfoques reparan el daño después de ocurrido. Además, la proteína huntingtina mutante tiene múltiples consecuencias deletéreas moleculares y celulares, cada una de las cuales podría ser la base de un enfoque terapéutico diferente. Sin embargo, la orientación individual puede ser insuficiente para un beneficio clínico significativo.
Huntingtina y la enfermedad de Huntington
El tratamiento de las afecciones neurodegenerativas es un reto no resuelto en la medicina moderna. Las enfermedades de Alzheimer, Parkinson, y esclerosis lateral amiotrófica, se manifiestan de forma frecuente como trastornos esporádicos; excepcionalmente los pacientes las heredan. Por otra parte, la enfermedad de Huntington (que implica la demencia, depresión, corea, y años de altos costos de atención) corresponde a una alteración autosómica dominante, causada por la expansión de una repetición en serie de CAG cerca del comienzo de la región codificadora en el gen de la proteína huntingtina. Los resultados de la mutación generan un ARN mensajero (ARNm) con más de 36 repeticiones y una proteína con más de 36 glutaminas en tándem. Eliminar el alelo mutante huntingtina por silenciamiento de ARN podría ser terapéutico, de hecho, un estudio realizado por Holly B. Kordasiewicz y colegas (Neuron 2012; 74:1031-44) es compatible con una estrategia para su puesta en práctica.
Se han descrito dos tecnologías de silenciamiento de ARN: el uso de oligonucleótidos antisentido y el ARN de interferencia. En el primero, regiones de ADN de un oligonucleótido antisentido se unen con el ARNm objetivo, y luego una enzima intracelular, la ARNasa H, escinde la cadena dúplex de ARN, degradando así el ARNm. El grupo de Holly B. Kordasiewicz utilizó oligonucleótidos antisentido para silenciar la huntingtina mutante en varios modelos de ratón para la enfermedad de Huntington. El tratamiento redujo los niveles de huntingtina mutante y mejoró una variedad de comportamientos aberrantes. Reducir la cantidad de huntingtina mutante fue un gran hallazgo. Además, el efecto saludable duró durante muchos meses después de haber dejado de aplicar los oligonucleótidos antisentido.
¿Cómo podrían 2 semanas de infusión de oligonucleótidos antisentido en líquido cefalorraquídeo (LCR) revertir o prevenir la afección durante tanto tiempo? Una posibilidad es que éstos sean resistentes y que los niveles terapéuticos duren meses. Sin embargo, la concentración de oligonucleótidos antisentido disminuyó mucho antes que los efectos saludables decayeran. Tal vez se requieran sólo unas pocas moléculas para reclutar la ARNasa H del ARNm mutante. Esto es una explicación intrigante para la terapia con oligonucleótidos antisentido y el ARN de interferencia. Ambos procesos pueden atacar libremente a muchas moléculas de ARNm en el tiempo.
Pero se podría proponer otra explicación (fig. 1). Si bien el ARNm de la huntingtina se expresa en el cerebro fetal, muchas veces la enfermedad de Huntington se vuelve clínicamente detectable entre los 30 y 40 años de edad. La huntingtina mutada (o su ARNm) preserva alguna función normal; los individuos que son homocigotos para el gen mutado no tienen una enfermedad más grave que aquellos con un único alelo mutante con la misma longitud de repetición de trinucleótidos CAG. Por lo tanto, los cambios producidos por la huntingtina mutante son sutiles y aumentan en el tiempo. Se cree que hay un efecto umbral. Antes de la aparición de la enfermedad, los sistemas de supervivencia celular mantienen el contenido celular de la huntingtina mutante por debajo del umbral para la enfermedad. Una vez que se supera, se produce la progresión de la condición. Así, en teoría, cualquier tratamiento que reduzca los niveles de ARN mutante lograría el mismo efecto terapéutico.
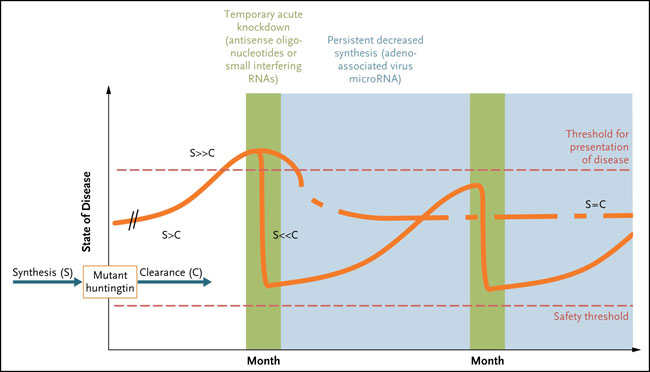
Esta investigación determinó que el tratamiento de ratones con la enfermedad de Huntington durante 2 semanas produce resultados terapéuticos durante varios meses. La observación es consistente con la idea de un efecto umbral para la acumulación de huntingtina mutante. La síntesis de la proteína mutada se produce en el útero, pero la presentación de la enfermedad a menudo comienza después de los 30 años de edad. Las neuronas eliminarían la huntingtina mutante y por tanto retrasarían la condición durante décadas. La acumulación de huntingtina mutante no es lineal, y parece que se acelera hasta un umbral antes de la presentación de la enfermedad. Los oligonucleótidos antisentido pueden reducir los niveles de síntesis a un punto que la neurona nuevamente elimine la proteína mutante. La caída excesiva puede reducir la cantidad total de huntingtina a niveles peligrosos, causando la disfunción neuronal por falta de suficiente huntingtina. Otra estrategia terapéutica consistiría en un efecto persistente y disminución de la síntesis, quizás mediante la administración de terapia génica: un virus adeno-asociados que exprese bajos niveles de ARN interferentes.
Incluso los estudios más importantes también tienen salvedades. Los modelos de ratón utilizados expresan altas cantidades de huntingtina mutada, de modo que el límite de huntingtina silenciada para establecer parámetros de seguridad no está todavía claro. Cabe destacar que el 75% del silenciamiento en ratones normales no dio lugar a cambios de conducta. No se sabe si este margen de seguridad es aplicable a seres humanos. Estudios neuropatológicos muestran menos encogimiento del cerebro en los animales tratados con oligonucleótidos antisentido. Sería útil conocer el tipo de células o tipos (neurona, neuroglia, microglia) que ocupan oligonucleótidos antisentido, así como el estado general de salud de las células cerebrales. La infusión de oligonucleótidos antisentido en el LCR de monos rhesus fue seguido por la absorción de compuestos según áreas de la corteza que son afectadas en los pacientes con enfermedad de Huntington, pero el cuerpo estriado (crítico en la enfermedad de Huntington) y otras regiones cerebrales muestran poca captación. Además, no se ha establecido el límite en seres humanos para el silenciamiento seguro de ambos alelos huntingtina. Podría ser más prudente eliminar sólo el alelo mutante, preservando la huntingtina normal, ya sea por terapia con oligonucleótidos antisentido o ARN de interferencia.
La entrega es el mayor problema para todas las aproximaciones de silenciación de genes, y los avances en el transporte de moléculas de la vasculatura a través de la barrera sangre-cerebro podrían facilitar el tratamiento de enfermedades cerebrales. Inyectar por vía intravenosa las microvesículas exosomas genera pequeños ARNs en el cerebro. Virus adeno-asociados administrados a la circulación pueden entrar en las neuronas y las células gliales y, con una ingeniería adecuada, podrían ocasionar a largo plazo la expresión de pequeños ARNs interferentes. El tratamiento más eficiente sería combinar el silenciamiento de la huntingtina en el cuerpo estriado (por medio de ARNs de interferencia viralmente entregados) y la administración periódica de oligonucleótidos antisentido en el LCR de la médula espinal.
Fuente bibliográfica
Hunting Down Huntingtin
Neil Aronin, M.D., and Melissa Moore, Ph.D.
RNA Therapeutics Institute and the Neurotherapeutics Institute, University of Massachusetts Medical School, Worcester (N.A., M.M.), and the Howard Hughes Medical Institute, Chevy Chase, MD (M.M.).
DOI: 10.1056/NEJMcibr1209595