Un antiguo fármaco para el Alzheimer
La forma más común de la enfermedad de enfermedad de Alzheimer (EA) se presenta esporádicamente a finales de la vida y se caracteriza por depósitos de ß-amiloide (Aß) en el cerebro. Los pacientes de inicio tardío producen péptidos Aß en niveles normales, pero tienen una capacidad limitada para eliminarlos desde el cerebro. Mayores concentraciones se asocian a perturbaciones de la función sináptica y la actividad de la red neuronal que probablemente subyacen al déficit cognitivo en la EA. Por otra parte, la acumulación de Aß genera una cascada patológica que finalmente lleva a la muerte neuronal. El factor de riesgo genético más influyente es la variación alélica de la apolipoproteína E (APOE), el alelo APOE4 aumenta dramáticamente el riesgo de enfermedad.
Los avances durante los últimos años nos han dado un conocimiento detallado de los mecanismos moleculares. Como la atención se ha centrado en el péptido beta amiloide, la hipótesis que prevalece es que las proteínas se agrupan en placas, dañando las células nerviosas del cerebro, provocando finalmente los síntomas característicos de la demencia senil. La teoría ha dado lugar a un gran número de potenciales fármacos que tratan de retrasar el desarrollo de la condición al disminuir la formación de, o incluso eliminar, las placas. Sin embargo, varios ensayos clínicos importantes han demostrado que este tipo de medicamentos no son en absoluto eficaces.
Éxito preclínico contra el Alzheimer
La enfermedad de Alzheimer es una condición neurodegenerativa crónica y devastadora que afecta la memoria del paciente, destruye la capacidad de razonar y hacer juicios racionales, y da lugar a una gran cantidad de síntomas conductuales y psiquiátricos. Con un nuevo caso cada 68 segundos en los Estados Unidos, las estimaciones indican que la prevalencia se podría triplicar a mediados de siglo, de 5,4 millones a 13,5 millones de pacientes. Si estas aproximaciones son correctas, la enfermedad generará una carga sin precedentes, médica, social y económica en nuestra sociedad.
Después de décadas de investigación sobre los mecanismos moleculares y genéticos subyacentes, es decepcionante que no existan terapias que permitan modificar la enfermedad en la práctica clínica. La gran cantidad de nuevos casos y el tiempo requerido para introducir un medicamento en el mercado aumentan la urgencia de identificación de dianas terapéuticas y nuevas estrategias. En un estudio reciente, Paige E. Cramer y colaboradores (Science 2012; 335:1503-6) informan que el bexaroteno, ya aprobado por la Food and Drug Administration para el tratamiento del linfoma cutáneo de células T, muestra una eficacia prometedora en modelos preclínicos.
Las lesiones características neuropatológicas de la enfermedad de Alzheimer son las placas neuríticas y los ovillos neurofibrilares. Las primeras constan de una pequeña agregación tipo péptido denominada β-amiloide (Aß), generada por escisión proteolítica de la proteína precursora de amiloide transmembrana (fig. 1). La gran mayoría de estos casos ocurre esporádicamente, a diferencia de la aparición temprana, la enfermedad familiar autosómica dominante, causada por mutaciones en uno de tres genes (amiloide que codifica la proteína precursora, la presenilina 1 o la presenilina 2), y se cree que es provocada por aumento de la producción o agregación de Aß. Los incidentes esporádicos serían el resultado de un fallo en los mecanismos de limpieza de Aß, y el principal factor de riesgo para estos casos es un polimorfismo que afecta a la apolipoproteína E (APOE), una proteína de transporte de colesterol que promueve la degradación proteolítica de Aß.
El gen que codifica la APOE es polimórfico, con la aparición de tres variantes alélicas en la población humana, APOE2, APOE3 y APOE4. El alelo más frecuente en la población general es APOE3, aunque el APOE4 está significativamente sobrerrepresentado en estos pacientes, lo que ocurre en el 40 y 65% de todas las personas afectadas. Aunque se lleve el alelo APOE4 no es ni necesario ni suficiente para la aparición de la afección, aunque su presencia incrementa significativamente el riesgo. Por el contrario, APOE2 es neuroprotector, y los portadores tienen una probabilidad reducida de enfermedad. Se desconoce el mecanismo por el cual APOE4 acelera la enfermedad, aunque tanto los efectos tóxicos de ganancia y pérdida de función (lo que podría comprometer la capacidad de APOE de eliminar a Aß) son posibles. La expresión de APOE se regula a través de la acción de los receptores nucleares del peroxisoma proliferador activado del receptor γ (PPAR-γ) y del receptor X del hígado en conjunto con el receptor X retinoide, lo que representaría una oportunidad para modular sus niveles farmacológicamente (fig. 1).
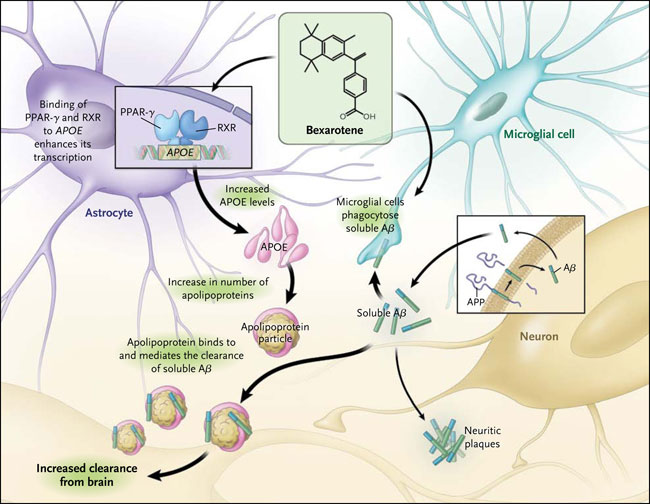
Las neuronas son la principal fuente celular que libera péptidos β-amiloide (Aβ), los cuales pueden formar placas neuríticas, una característica patológica del Alzheimer. La apolipoproteína E (APOE) es liberada principalmente por astrocitos y la microglia, aunque una pequeña cantidad también proviene de las neuronas. La APOE se asocia con lipoproteínas para formar partículas de lipoproteínas asociadas a APOE, las cuales se unen a Aß solubles, promoviendo su limpieza desde el cerebro. Este artículo informa que el bexaroteno, un agonista de los receptores nucleares, aumenta la expresión de APOE, que estimula la eliminación de Aß y mejora los déficits cognitivos en un modelo murino. PPAR-γ denota el receptor activado por proliferadores de peroxisomas γ, y RXR el receptor X retinoide.
P. Cramer y colegas plantearon la hipótesis que el bexaroteno, un agonista del receptor X retinoide, podría revertir el fenotipo degenerativo en ratones transgénicos para la enfermedad de Alzheimer mediante la mejora de los niveles de APOE, reduciendo así la cantidad de Aß cerebral. Esto es exactamente lo que ocurrió en los animales, en donde la rapidez de la limpieza fue sorprendente, con áreas de placas Aß reducidas en más del 50% dentro de las 72 horas. El tratamiento también invirtió los déficits cognitivos, sociales y olfativos. Es importante destacar que el bexaroteno disminuyó los niveles de Aß de manera dependiente de APOE (es decir, que no tuvo ningún efecto en ratones sin APOE). Los autores encontraron que la droga fue capaz de convertir la microglia en su estado de activación alternativa y por lo tanto promover la fagocitosis Aß.
Rara vez la ciencia es sencilla. El bexaroteno redujo rápidamente las placas en un 75% después de 14 días en los ratones transgénicos, pero el tratamiento en el transcurso de 3 meses estuvo acompañado por una reversión de la placa hasta el punto en el que era equivalente a la de los animales control. Las implicaciones clínicas de esta observación no están todavía claras, ya que pueden indicar que el régimen de tratamiento o el intervalo de administración deben ser cuidadosamente considerados.
A pesar de los alentadores resultados, uno no podría dejar pensar que el campo ha pasado por esto antes, ya que los éxitos en modelos preclínicos hasta ahora no han podido ser trasladados a la clínica.
¿Por qué las terapias e intervenciones para la enfermedad de Alzheimer han tenido éxito en modelos preclínicos, pero no cuando se han evaluado en seres humanos? La respuesta a esta pregunta es multifactorial, pero la explicación más parsimoniosa es que la mayoría de los compuestos son evaluados en modelos que albergan patología amiloide única y, en particular, carecen de otras características críticas patológicas, tales como los ovillos neurofibrilares y muerte neuronal. Otra razón probable es que un trastorno tan complejo como el Alzheimer requiere múltiples intervenciones terapéuticas. Sólo un ensayo clínico bien diseñado y ejecutado cuidadosamente revelará si esta clase de medicamentos hace honor a sus expectativas. Hasta que estos trabajos se lleven a cabo, sería un error ofrecer tales tratamientos a pacientes con Alzheimer.
Fuente bibliográfica
Preclinical Success against Alzheimer’s Disease with an Old Drug
Frank M. LaFerla, Ph.D.
Institute for Memory Impairments and Neurological Disorders, University of California, Irvine
N Engl J Med 2012; 367:570-572