El camino del Alzheimer por el cerebro
La enfermedad de Alzheimer se caracteriza neuropatológicamente por la presencia de placas beta-amiloide y de ovillos neurofibrilares, que están formados por agregados fibrilares y formas hiperfosforiladas asociadas a los microtúbulos de la proteína tau. Sus primeras etapas muestran la acumulación anormal de tau en la corteza entorrinal mientras que los estadios posteriores se observa su acumulación en el hipocampo seguido hacia las áreas neocorticales. Una de las cuestiones más intrigantes y poco exploradas es si la patología y/o disfunción de la corteza entorrinal es la que inicia la progresión anatómica de la enfermedad, o si la patología y/o disfunción en áreas extra-hipocampales se desarrolla de forma independiente, y no está relacionada con eventos que ocurren en la zona entorrinal. Hay una serie de interesantes observaciones, pero circunstanciales, que apoyan la hipótesis de la propagación trans-sináptica para el Alzheimer, tanto en términos de desarrollo patológico como para el resultado funcional.
Propagación de enfermedades neurodegenerativas
Los médicos que atienden a pacientes con enfermedades neurodegenerativas a menudo creen que las patologías se extienden a través de sus cerebros. Los estadios anatómicos de Braak de las enfermedades de Alzheimer y de Parkinson reflejan la opinión de que las características patológicas de estas condiciones son capaces de propagarse. Por supuesto, sus impresiones clínicas y anatomías distintivas podrían explicarse de muchas maneras. Por ejemplo, podrían resultar de la vulnerabilidad selectiva frente a la creciente presión metabólica externa de las neuronas, reflejar el hecho que el daño neuronal provenga de otros lugares adyacentes, producto de cierta difusión de proteínas patológicas desde las neuronas dañadas hacia neuronas contiguas, o ser cualquier combinación de estos factores.
Hasta hace poco, sólo en la enfermedad por priones se había demostrado tal propagación patológica, probando que el agente de proliferación es la proteína del prión. Recientemente, sin embargo, se ha planteado la idea que otras alteraciones neurodegenerativas podrían propagarse a través del movimiento de las proteínas, como en los casos de beta-amiloide (Aβ), tau y α-sinucleína. Las unidades aberrantes Aβ y tau están implicadas en la enfermedad de Alzheimer, la tau mutante en la demencia fronto-temporal y la α-sinucleína mutante causa la enfermedad de Parkinson.
La primera evidencia que otras proteínas podrían ser patológicas fue descrita en 1994, cuando la introducción de tejido cerebral de un paciente con enfermedad de Alzheimer en cerebros de monos adultos generó la aparición de placas Aβ. Sin embargo, este informe fue ignorado, en parte porque el trabajo en primates de edad avanzada es difícil y lento. Mayor interés se dio por los experimentos con ratones transgénicos que muestran que la enfermedad por Aβ podría acelerarse en estos animales (que son genéticamente susceptibles a enfermedades como el Alzheimer) a través de inoculaciones de extractos de cerebro de un paciente con Alzheimer (inyecciones intraperitoneales de fracciones cerebrales enriquecidas con Aβ también potenciaron una neuropatología por Aβ). Debido a que la amiloidosis cerebral Aβ en ratones susceptibles se puede inducir por pequeñas cantidades extractos cerebrales ricos en Aβ, también se ha planteado la posibilidad teórica de transmitir actividad amiloide Aβ a pacientes mediante instrumentos neuroquirúrgicos contaminados. Además, se sabe que los enfermos con Parkinson sobrevivientes a injertos de células dopaminérgicas fetales durante más de 10 años poseen α-sinucleína a la forma de cuerpos de Lewy en el tejido injertado.
Recientemente, los grupos investigadores de Li Liu (PLoS One 2012; 7(2):e31302) y Alix de Calignon (Neuron 2012; 73:685-97) encontraron que las formaciones tipo ovillos característicos del Alzheimer podrían propagarse por vía transináptica a través de las neuronas de la corteza entorrinal en ratones transgénicos que expresan una tau mutante. Liu y colegas construyeron una tau humana alterada, para que se "activara" y funcionara sólo en neuronas corticales de la corteza entorrinal de ratones jóvenes, pero a medida que estos envejecían, la condición se propagaba al subículo y a las áreas conectadas sinápticamente en el hipocampo y corteza, aunque las neuronas de las últimas zonas tenía una expresión extremadamente baja de la proteína mutante. De Calignon y colaboradores presentaron un enfoque muy parecido y tuvieron resultados similares, pero señalaron que los depósitos de tau en las neuronas descendentes también contenían la proteína endógena. Este hallazgo sugiere que los depósitos humanos de tau habrían reclutado a la proteína del ratón en los depósitos. Ambos grupos de investigadores informaron que, aunque la propagación de la enfermedad fue coherente con la propagación mono-sináptica secuencial, no todas las poblaciones de neuronas estuvieron igualmente afectadas.
Los estudios en humanos y animales han sido complementados con ensayos celulares, tanto tau y α-sinucleína mal plegadas pueden ser captadas por las células e inducir el mal plegamiento de la proteína endógena. Las últimas observaciones abren camino a experimentos mecanicistas sobre la naturaleza de los fenómenos implicados (fig. 1).
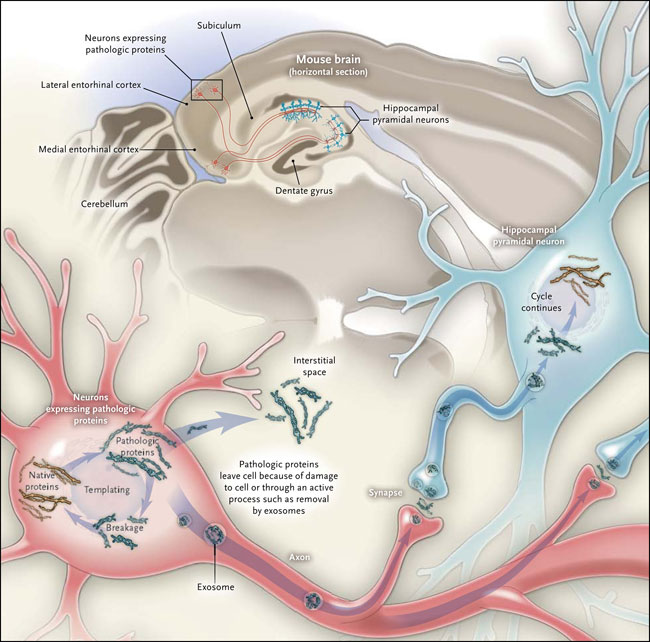
Un ciclo de plantillas de una proteína patológica, junto con la rotura y la transferencia entre células, puede conducir a la propagación de la enfermedad, por ejemplo, entre neuronas adyacentes dentro de una vía. Dos nuevos estudios apoyan la hipótesis que la enfermedad de Alzheimer y la demencia fronto-temporal se extienden de esta manera.
Colectivamente, estos estudios, apoyan la hipótesis que la propagación patológica es un fenómeno general de las enfermedades neurodegenerativas, representando un destacado aspecto para la biología y epidemiología de la enfermedad de Alzheimer, el Parkinson y la demencia fronto-temporal, así como respecto a nuevos enfoques terapéuticos. Ahora, no está claro si los procesos de difusión dentro del cerebro son fisiológicos en lugar de acontecimientos estrictamente patológicos. El proceso de propagación sugiere que hay zonas en los que las terapias se podrían centrar. También implica que la enfermedad estocástica puede tener su origen a partir de una sola célula.
A pesar de la emocionante naturaleza de estos estudios, se aconseja precaución en el uso de las palabras "prión" o "prionoide" o "tipo prión". Las enfermedades priónicas pueden propagarse de animal a animal, de animal a humano y de persona a persona. También sabemos que el agente prión infeccioso es estable en el medio ambiente durante muchos años. No existe ninguna evidencia de que nada de esto sea cierto para la enfermedad de Alzheimer, Parkinson o la demencia fronto-temporal y sus agentes etiológicos, y se debe evitar las implicaciones que conlleven la palabra "prión". Finalmente, parece que estas enfermedades no parecen compartir un fenómeno en común con las enfermedades priónicas.
Fuente bibliográfica
The Spread of Neurodegenerative Disease
John Hardy, Ph.D., and Tamas Revesz, M.D.
Reta Lila Weston Laboratories and the Department of Molecular Neuroscience, University College London Institute of Neurology, Queen Square, London
N Engl J Med 2012; 366:2126-2128