El melanoma se resiste…
La droga experimental PLX4032, es un inhibidor selectivo de la enzima quinasa B-RAF, ha representado una significativa promesa en los primeros ensayos clínicos contra el melanoma maligno. Sin embargo, algunos pacientes desarrollan resistencia a este fármaco en una etapa temprana del tratamiento. Descubrir los mecanismos mediante los cuales se produce la resistencia a PLX4032 es un paso importante hacia el desarrollo de terapias más eficaces para esta enfermedad.
Recientemente, grupos de investigadores han publicado algunos trabajos sobre los mecanismos que supuestamente confieren el desarrollo de dicha resistencia al fármaco.

La resistencia a la inhibición BRAF en el melanoma
Aproximadamente el 50% de los melanomas contienen una mutación en el gen que codifica a BRAF, una proteína quinasa de la familia RAF que fosforila la proteína MEK y activa la vía de señalización ERK. Esta alteración, que en la mayoría de los casos es la sustitución del ácido glutámico por valina en la posición 600 de la proteína, activa y desregula la actividad de la quinasa de BRAF. Los inhibidores selectivos de RAF, como el PLX4032, tienen una notable actividad clínica en pacientes con melanomas que contienen la mutación V600E en BRAF. Sin embargo, siguiendo el patrón de otros inhibidores de quinasas oncogénicas, las respuestas a PLX4032 a menudo son profundas, pero temporales. Dos estudios recientes, uno realizado por Ramin Nazarian y colegas (Nature 2010; 468:973-977) y otro por Cory M. Johannessen y colaboradores (Nature 2010; 468:968-972), arrojan nuevas evidencias sobre algunos de los mecanismos de resistencia a los inhibidores RAF y, por tanto, sugieren estrategias terapéuticas para estos pacientes.
Entender los mecanismos de sensibilidad y la adquisición de resistencia de los melanomas con mutación BRAF V600E a los inhibidores de RAF requiere un conocimiento básico de la forma de acción de estos fármacos. La activación de las proteínas RAS estimula las quinasas RAF, tales como ARAF, BRAF y RAF1 (o CRAF). Este proceso provoca la fosforilación de las quinasas MEK que, a su vez, fosforilan las quinasas ERK (figura 1). La activación de ERK regula múltiples procesos celulares claves que se requieren para la proliferación celular y la supervivencia. RAS activa a RAF mediante su localización en la membrana plasmática y facilita su dimerización. Como es de esperar, los inhibidores de MEK suprimen la señalización de ERK en todas las células normales y tumorales. En contraste, PLX4032 inhibe la vía de ERK y la proliferación celular sólo en los tumores con mutantes BRAF. En neoplasias y células normales con RAF tipo salvaje, PLX4032 provoca la paradójica activación de la vía. Esta inhibición tumoral específica de la señalización de ERK por PLX4032 probablemente es la base de su amplio espectro terapéutico, permitiendo una inhibición más potente de la vía que lo logrado con inhibidores de MEK, que están limitados por los efectos tóxicos de la inhibición en las células normales.
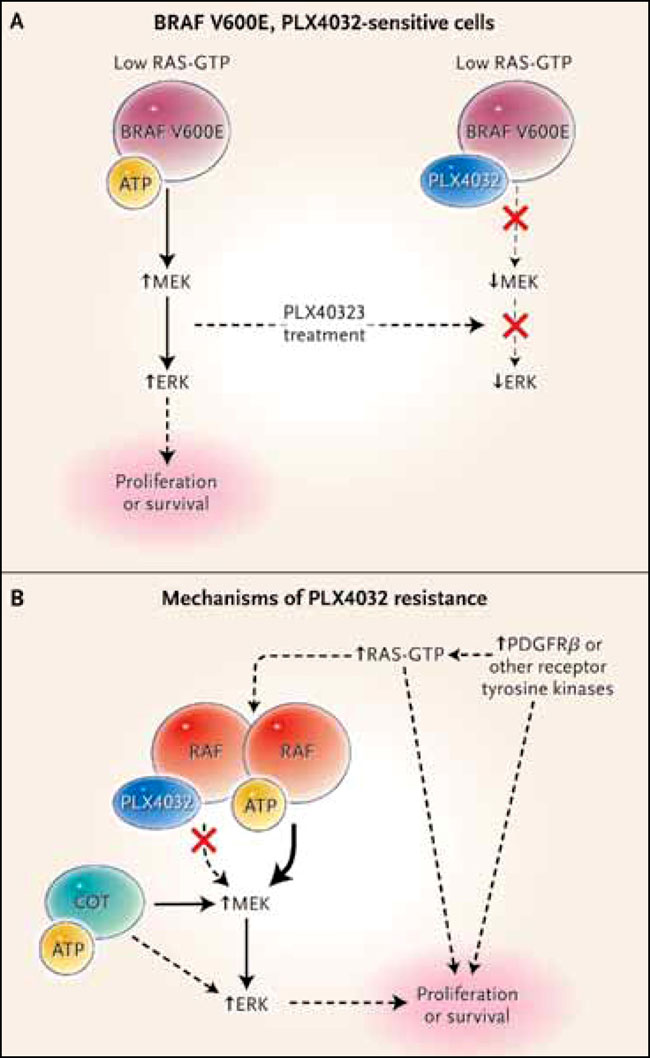
En el panel A, cuando la actividad RAS es baja en las células del melanoma que expresan la mutación BRAF V600E, los niveles de dímeros de RAF son bajos, y los inhibidores RAF se unen, los monómeros inhiben a RAF y, por tanto la actividad de MEK y ERK. En el panel B, la sobreexpresión de RAF1 o la activación de RAS como resultado de la mutación RAS o la sobre-activación de la tirosina quinasa del receptor, promueve la formación de dímeros de RAF. En células que expresan dímeros RAF, la unión de inhibidores de RAF a un miembro del dímero transactiva el otro. En estas células, PLX4032 no inhibe la señalización de la quinasa MAP, conduciendo resistencia a los medicamentos. Por otra parte, la sobreexpresión de MAP3K8 (o COT) da lugar a la activación de MEK y ERK independiente de RAF, y por lo tanto resistencia a PLX4032. La activación del receptor tirosina-quinasa también puede causar resistencia a PLX4032 a través de la activación de RAS, o por la activación de vías de señalización paralelas, disminuyendo la dependencia celular de la señalización RAF.
¿Por qué PLX4032 inhibe la activación de RAF sólo en células con la mutación BRAF V600E? En células con actividad RAS adecuada, las proteínas RAF forman dímeros, y la unión de la droga a un RAF en el dímero activa al otro. En los melanomas con la mutación BRAF V600E, los niveles de RAS activos son demasiado bajos para promover la adecuada formación de dímeros RAF, y PLX4032 inhibe la actividad de RAF y la señalización de ERK (figura 1). Este modelo es consistente con la observación de que la introducción de mutantes RAS (activados) en células con mutantes BRAF causa insensibilidad de la vía ERK a la droga. Este modelo sugiere que el aumento de la dimerización de RAF (debido a la activación de RAS o al aumento de la expresión MAR) hará que la señalización de ERK sea insensible a PLX4032.
La relevancia clínica de este modelo ha sido confirmada por el grupo de R. Nazarian, quienes describen a un paciente en el que la resistencia a PLX4032 se da a través de una mutación en NRAS después del tratamiento. Así, el mecanismo subyacente a la inhibición selectiva de la señalización ERK en células mutantes BRAF por PLX4032 es también una especie de “talón de Aquiles”. La resistencia a los inhibidores del receptor del factor de crecimiento epidérmico, en tumores Kit y Abl, en donde estas quinasas se activan es a menudo el resultado de mutaciones denominadas “guardianes” (gatekeepers) que impiden la unión del fármaco a la enzima. Sorprendentemente, a pesar que tales alteraciones confieren resistencia a PLX4032 en modelos de laboratorio, esto no se observó en los 16 tumores resistentes analizados por R. Nazarian y colaboradores.
Los resultados de Cory M. Johannessen y colegas sugieren otro mecanismo para la resistencia de la señalización ERK a la inhibición RAF en las células que consideraría el efecto de las mutaciones BRAF V600E. Estos investigadores usaron una nueva técnica - la introducción de una colección de librerías de ADN, codificantes cada una de ellas de una quinasa diferente – que aplicaron sobre células tumorales con mutación BRAF V600E para la detección de quinasas que confieren resistencia a la inhibición de RAF. Usando esta forma, ellos confirmaron un hallazgo anterior: que la sobreexpresión de RAF1 confiere resistencia a la inhibición de RAF. Asimismo, pusieron de manifiesto que la sobreexpresión de la proteína activada por mitógeno 8 (MAP3K8 o COT), que fosforila a MEK de manera independiente de RAF, también pueden mediar la resistencia a los inhibidores de RAF. Aunque se esperaría que tales células conserven la sensibilidad a un inhibidor de MEK, los datos sugieren que la activación COT también omite a MEK y activa directamente a ERK (figura 1B).
El estudio de R. Nazarian propone una tercera posibilidad para la resistencia adquirida, en la que la activación de otras vías hace que la célula del tumor sea menos dependiente de la señalización de ERK. En estos tumores, la activación de ERK sigue siendo sensible a los inhibidores de RAF. En concreto, se señala que el crecimiento derivado del receptor del factor de crecimiento derivado de plaquetas β (PDGFRβ), un receptor de tirosina quinasa, se sobre-expresa en modelos celulares seleccionados para la resistencia al inhibidor RAF en cultivos celulares y en un subgrupo de muestras de biopsias obtenidas de pacientes con tumores progresivos. En líneas celulares, la sobreexpresión de PDGFRβ se asoció con resistencia a los efectos antiproliferativos de los inhibidores RAF, a pesar de la inhibición de la señalización de ERK en presencia de la droga.
Estos estudios sugieren nuevas estrategias para tratar a los pacientes con melanomas, portadores de la mutación BRAF V600E y en los que se desarrolla la resistencia a PLX4032. La resistencia causada por la activación de COT o PDGFRβ debería ser controlada por los inhibidores de estas quinasas en combinación con el inhibidor de RAF. La resistencia generada por mutaciones que afectan a las proteínas RAS podría ser más difícil de tratar debido a que los intentos por desarrollar inhibidores directos de RAS han fracasado. La inhibición combinada de MEK y RAF puede resultar útil en estos individuos, pero no afectaría a la activación de otros objetivos RAS. Por último, hay que destacar que han sido analizados muy pocos tumores resistentes. Se necesitan estudios adicionales para determinar la prevalencia relativa de cada uno de estos eventos y precisar si otros mecanismos de resistencia a inhibidores de RAF evitan el bloqueo de la señalización de ERK o hacen que el tumor crezca de manera independiente a ERK.
Fuente bibliográfica
Resistance to BRAF Inhibition in Melanomas
David B. Solit, M.D., and Neal Rosen, M.D., Ph.D.
From the Departments of Medicine (D.B.S., N.R.) and Molecular Pharmacology and Chemistry (N.R.), Memorial Sloan-Kettering Cancer Center, New York.
N Engl J Med 2011; 364:772-774