Nefropatía diabética, por culpa de los podocitos
La nefropatía diabética, también conocida como síndrome de Kimmelstiel-Wilson, o glomeruloesclerosis nodular diabética y glomerulonefritis intercapilar, es una enfermedad progresiva del riñón causada por la angiopatía de los capilares en los glomérulos renales. Se caracteriza por la presencia de síndrome nefrótico y glomeruloesclerosis difusa, debido principalmente a la diabetes mellitus de larga data. Es la principal causa de insuficiencia renal crónica en los Estados Unidos y en otras sociedades occidentales. También representa una de las complicaciones más importantes en términos de morbilidad y mortalidad en los pacientes diabéticos.
En los últimos años, las alteraciones estructurales del podocito han sido consideradas como posibles contribuyentes a la patogénesis de la nefropatía diabética. Estas células son parte integral de la barrera de filtración, y se han observado cambios en su estructura en una amplia gama de enfermedades glomerulares proteinúricas, como la diabetes. Contrario a otras células del glomérulo, los podocitos tienen una capacidad limitada para replicarse, por lo menos después del nacimiento. Por lo tanto, necesariamente se requeriría que las células residuales cubran un área más extensa de la membrana basal glomerular. Esto podría causar la pérdida de procesos bases y reducir la capacidad de los podocitos a permanecer unidos a la membrana, con consecuentes áreas al descubierto, las que serían potenciales puntos de partida de la glomeruloesclerosis.
Proteinuria, podocito y resistencia a la insulina
La nefropatía diabética es una enfermedad crónica progresiva que afecta entre un 20 y un 40% de los pacientes con diabetes mellitus. Los ensayos clínicos han demostrado que el control estricto de la hiperglucemia y la hipertensión pueden ralentizar su progresión y que la resistencia a la insulina se correlaciona con la aparición y gravedad de la albuminuria. Esta correlación también se ha encontrado en personas normotensas que no tienen diabetes, lo que sugiere que la resistencia a la insulina por sí misma puede causar albuminuria. Sobre la base de estas observaciones, Gavin I. Welsh y colegas (Cell Metab 2010; 12:329-340) investigaron el papel de la señalización de la insulina en el riñón y concluyeron que la deficiencia de los receptores de insulina en los podocitos conduce a una lesión glomerular parecida a la nefropatía diabética, pero en ausencia de hiperglucemia.
La insulina es el principal regulador del metabolismo de la glucosa y de los lípidos, una función que depende esencialmente de su interacción con el receptor de la insulina, expresada principalmente en el músculo, grasa, hígado y cerebro. El riñón no ha sido tradicionalmente considerado como un órgano blanco de la acción insulínica, aunque se sugiere que los sensibilizadores de insulina pueden ser superiores a otros agentes hipoglucemiantes a la hora de conferir protección contra la nefropatía diabética. Después de describir una vía de señalización para una insulina funcionalmente intacta en podocitos, el grupo de Gavin I. Welsh ahora demuestra un efecto directo de la señalización del receptor de insulina sobre la función de los podocitos in vitro e in vivo.
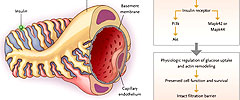
Los podocitos son células altamente especializadas de los glomérulos del riñón que ayudan a impedir la proteinuria mediante una reglamentación compleja del citoesqueleto de actina en sus procesos bases. Los investigadores diseñaron dos modelos de ratones en los que el gen que codifica el receptor de la insulina (Insr) podría ser eliminado de una manera específica en los podocitos. En los animales en los que se eliminó el gen, se desarrolló albuminuria, junto con la supresión de los procesos bases de los podocitos, la apoptosis, el engrosamiento de la membrana basal glomerular, y el aumento de la glomeruloesclerosis - todas características histológicas típicas de la nefropatía diabética.
Análisis de glomérulos aislados de ratones tipo salvaje y de ratones con deleción del receptor de la insulina en podocitos que fueron tratados con insulina mostraron que la señalización insulínica en los podocitos se produce principalmente por dos vías: una involucra a PI3K (fosfatidilinositol 3-quinasa) y Akt (homologo viral 1 del oncogene del timoma murino de v-akt) y la otra a Mapk42 o Mapk44 (proteína quinasa activada por mitógenos 42 ó 44) (figura 1). Los autores también determinaron que la insulina induce directamente la reorganización a corto plazo del citoesqueleto de actina de podocitos humanos in vitro, provocando la retracción de los procesos celulares y aumento de la permeabilidad. Estos hallazgos son consistentes con la evidencia clínica que la infusión de la insulina en personas normales genera proteinuria transitoria. El cómo estas vías interactúan para causar nefropatía diabética está aún por establecerse.
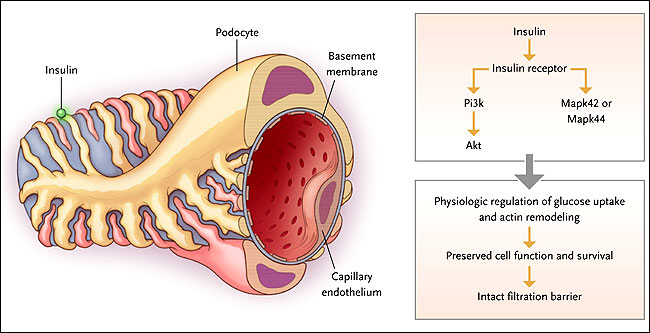
El presente estudio indica que, en podocitos de ratones de tipo salvaje, la activación de insulina a partir del receptor de insulina resulta en la fosforilación de Akt y Mapk42 o Mapk44, generando la remodelación fisiológica del citoesqueleto de actina y la preservación de la función celular y la supervivencia. En los podocitos de los ratones carentes del receptor de insulina, la señalización de la hormona a través de Akt y Mapk42 o Mapk44 es suprimida y resulta en la eliminación de los procesos bases, engrosamiento de la membrana basal glomerular, y el mal funcionamiento o muerte celular, lo que lleva a la proteinuria.
Puesto que la estimulación sin oposición de la vía MAPK por medio de la hiperinsulinemia compensatoria se produce en pacientes con resistencia a la insulina, se podría argumentar que el fenotipo de la enfermedad en ratones mutantes puede ser más representativo de la deficiencia insulínica que de la resistencia a la insulina. Un hallazgo clave de esta investigación fue la marcada variabilidad en la severidad del fenotipo renal de los ratones mutantes, de acuerdo con la evidencia clínica que la nefropatía diabética no se desarrolla en todos los pacientes con receptor de la insulina mutante. El hallazgo que la nefropatía diabética se desarrolló sólo en una de las dos hermanas que llevaban la misma mutación en INSR sugiere que las alteraciones que afectan al receptor de la hormona pueden ser insuficientes para provocar un fenotipo patológico renal en humanos.
Pero ¿por qué la señalización de la insulina es importante para la función de los podocitos? Imágenes del metabolismo de la glucosa in vivo han demostrado que el cerebro, músculo esquelético y la grasa son los principales implicados en la captación de glucosa, mientras que el parénquima renal requiere grandes cantidades sólo bajo condiciones patológicas, con sólo una mínima recaptación fisiológica renal debido a la excreción de glucosa. Es posible que el receptor de la insulina de sus podocitos sirva sobre todo como un receptor de proteínas implicadas en la remodelación del citoesqueleto de actina y en la orientación de los procesos bases. El hecho que la nefrina, un regulador clave de la permeabilidad de los podocitos, sea esencial para señalización de la insulina en los podocitos admite esta posibilidad.
El receptor insulínico en sus podocitos es por lo tanto un objetivo atractivo para el desarrollo de nuevos agentes antiproteinúricos. El trabajo de Gavin I. Welsh y colegas requiere la realización de más estudios experimentales que puedan caracterizar los mecanismos por el cual la insulina y el receptor de la insulina afectan la función del podocito y, a su vez, los ensayos clínicos aleatorios puedan abordar el papel de los sensibilizadores de insulina en el tratamiento y prevención de la nefropatía diabética y de otras enfermedades glomerulares.
Fuente bibliográfica
Proteinuria, the Podocyte, and Insulin Resistance
Alessia Fornoni, M.D., Ph.D.
From the Division of Nephrology and Hypertension and the Diabetes Research Institute, University of Miami Miller School of Medicine, Miami.
N Engl J Med 2010; 363(21):2068-2069