Mejorando la visión en la retinitis pigmentaria
A nivel mundial, existen más de 2 millones de personas afectadas por un conjunto de enfermedades agrupadas bajo la denominación de retinosis pigmentaria. Se trata de una forma hereditaria de degeneración de la retina caracterizada por la pérdida progresiva de la visión hasta terminar en una ceguera total.
La enfermedad es el resultado de diversas mutaciones en más de 44 genes expresados en los fotorreceptores, los cuales luego degeneran, causando la pérdida de la visión nocturna. Posteriormente, los conos, células responsables del color y la agudeza visual durante el día, pierden progresivamente sus segmentos fotorreceptivos externos, lo que lleva a la total ceguera. A pesar de esta pérdida de sensibilidad, los conos permanecen durante mucho tiempo en los seres humanos y animales, pero no se sabe si pueden ser reactivados o si su información puede fluir a través de los circuitos visuales durante una ventana de tiempo considerable después de la pérdida de fotosensibilidad.
Hasta ahora no se advertía concretamente si una intervención terapéutica sería capaz de restaurar dichos conos insensibles.

Posibles tratamientos de la retinitis pigmentaria
La retinitis pigmentaria, un término acuñado por el oftalmólogo holandés Franciscus Cornelis Donders hace más de 150 años, integra un grupo de enfermedades crónicas degenerativas de la retina de carácter genético. La retina es una estructura compleja que recubre la superficie interna del ojo y es parte del sistema nervioso central. Las personas que la padecen generalmente pierden la visión nocturna en un grado mayor de lo que pierden la visión diurna y pierden la visión lateral (periférica) antes de perder la de la lectura (central). Decenas de cambios genéticos y una multitud de mecanismos conducen a la afección.
Se han descrito cuatro enfoques principales para su tratamiento, cada uno de los cuales se adapta a una o a varias etapas de la enfermedad (figura 1). El primer esquema consiste en la corrección de las alteraciones bioquímicas que provocan un componente de pérdida visual mientras que algunos fotorreceptores permanecen estructuralmente intactos. Un ejemplo es la corrección de una anomalía del ciclo visual (retinoides) en la forma grave de la retinitis pigmentaria conocida como amaurosis congénita de Leber, específicamente, el subtipo causado por mutaciones en el gen RPE65. La terapia génica subretinal, en los que se inserta un gen RPE65 normal debajo de la retina mediante inyección intraocular, corrige con éxito y seguridad la disfunción del ciclo visual en los pacientes. El mecanismo subyacente de la amaurosis congénita de Leber es complejo, porque también hay una degeneración progresiva de la retina. Se desconoce si la corrección del defecto ciclo-visual detiene o ralentiza esta degeneración. Otro ejemplo, es la administración de retinoides orales para lograr eludir farmacológicamente el ciclo visual.
Un segundo enfoque para el tratamiento de la retinosis pigmentaria está dirigido a detener o retardar la degeneración progresiva de los fotorreceptores. Incluye el uso de suplementos nutricionales, factores neurotróficos, y otros productos farmacéuticos que mejoran la viabilidad de las neuronas mediante la inhibición de las vías proapoptóticos, la activación de la señalización antiapoptótica, la reducción de la producción de moléculas retinotoxicas y limitar el daño oxidativo.
Dos enfoques terapéuticos son apropiados para los pacientes con estadios avanzados de la retinosis pigmentaria, en los que hay poco o nada de fotorreceptores funcionales. Uno tiene como objetivo regenerar los fotorreceptores perdidos (o mal desarrollados) mediante el trasplante o la manipulación genética de células de la retina no fotorreceptoras, como la glía. El otro implica la creación de señales eléctricas en la vía visual para sustituir la entrada habitual de los fotorreceptores. Los implantes electrónicos de retina son un ejemplo, y los ensayos clínicos de estos dispositivos están en curso. Volker Busskamp y colegas (Science 2010; 329:413-417) acaban de describir una nueva estrategia para esta cuarta categoría. Se utilizó halorrodopsina de arqueobacterias, un canal activado por la luz, para resintetizar genéticamente células fotorreceptoras de cono remanentes en fases muy tardías de la enfermedad a través de un modelo de ratón para la degeneración de la retina - cuando los fotorreceptores y la mayoría de los conos se han perdido. Este planteamiento "optogenético" fue exitoso para la transferencia de información sobre los cambios en la intensidad de luz desde la retina hacia las células ganglionares y el cerebro visual.
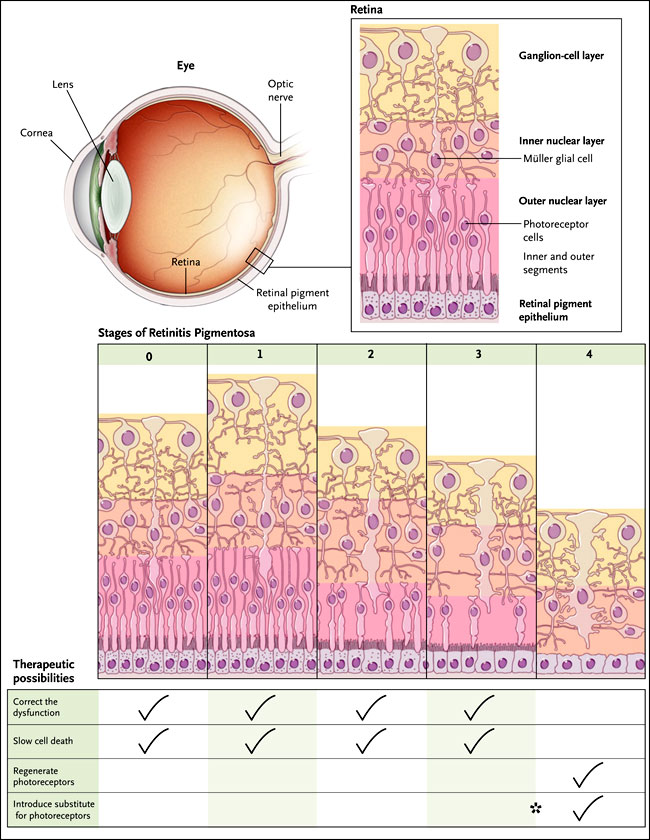
Los cambios en la arquitectura de la retina observados durante la progresión de la retinitis pigmentaria pueden dividirse en cinco etapas. La etapa 0 representa la arquitectura normal presente en una región de la retina en algunos pacientes con retinitis pigmentaria, por ejemplo, el área central de la retina puede tener la estructura y función normal a pesar de las anomalías en varios lugares de la retina periférica. La etapa 1 representa la primera alteración detectable: una retina engrosada con laminación y función normal. La enfermedad se vuelve clínicamente manifiesta con la pérdida de núcleos de los fotorreceptores, el acortamiento de los segmentos internos y externos de los fotorreceptores, y la despigmentación del epitelio pigmentario de la retina (etapas 2 y 3). La etapa más avanzada de la enfermedad (fase 4) se caracteriza por una retina adelgazada, en el que hay una importante mortalidad de células neuronales y pérdida de la arquitectura laminar con gliosis. Se ha descrito una compleja reorganización de los circuitos de la retina en esta etapa y en etapas anteriores. El esquema señala cuatro categorías respecto a las actuales posibilidades terapéuticas, con marcas de verificación que indican las etapas de la enfermedad durante la cual pueden ser más aplicadas. Entre los métodos que sustituyen la función de los fotorreceptores normales está el uso de herramientas optogenéticas para resintetizar "a ciegas" los conos, recientemente descrito por la investigación del grupo de V. Busskamp. Este enfoque puede aplicarse a la retina que tiene núcleos de fotorreceptores remanentes pero sin segmentos externos, lo que refleja un punto en la progresión de la enfermedad que está entre las etapas 3 y 4 (asterisco). Las etiquetas en negrita en la tabla (superior derecha) representan las tres capas nucleares y el pigmento de la capa epitelial de la retina.
¿Cuáles son los obstáculos que se podrían encontrar entre este estudio y la clínica? El primero es la necesidad de determinar la proporción de pacientes con retinitis pigmentaria que tienen algunos conos pero carecen de visión. Este impedimento puede ser eliminado mediante la medición colocalizada de la visión y la arquitectura de la retina de corte transversal con el uso de métodos actualmente disponibles. Si resulta que los candidatos apropiados son una minoría muy pequeña de la población, el número para la terapia se podría aumentar mediante la modificación del enfoque hacia las neuronas dianas alternativas, como las células bipolares y células ganglionares.
Un segundo obstáculo es la posible fototoxicidad asociada con el requisito de la alta irradiación de los canales activados por luz. Los límites de fototoxicidad para el epitelio pigmentario de la retina normal se han recalibrado, y el epitelio pigmentario de la retina enferma en los pacientes con retinitis pigmentaria en etapa avanzada puede ser aún más sensible a la toxicidad de la luz que el epitelio pigmentario de la retina normal. Por lo tanto, se justifican más investigaciones para el desarrollo de nuevos canales con mayor sensibilidad a la luz.
Un tercer obstáculo es el desarrollo de medios para estimular la óptica, con un control preciso del espacio-tiempo, de las regiones de la retina que han recibido tratamiento. Hasta que los canales puedan ser diseñados de forma que sean activados con luz natural, el entorno visual tiene que ser transformado electrónicamente usando una señal de vídeo, que se procesa para volver a mostrar los resultados, y así coincidir con el rango de radiación óptima de los canales. El medio ambiente transformado se puede presentar en forma pasiva bajo el supuesto de que los pacientes puedan dirigir su mirada hacia la señal visual. Sin embargo, puede no ser viable, porque muchos pacientes con pérdida profunda de la visión carecen del control de la mirada oculomotora dirigida. Por otra parte, el mundo natural procesado se puede presentar activamente en la retina tratada. A tal fin, los sistemas de proyección ocular con una función de seguimiento del ojo tienen que ser optimizados en estas personas con gran pérdida de la visión.
A pesar de los grandes avances científicos en la comprensión de la retinosis pigmentaria desde la época de Donders, las terapias basadas en la ciencia están limitadas en número.
Cuando el progreso terapéutico se obtiene a partir de modelos animales para la condición oftalmológica, este también debería sustentar la realización, dentro de un plazo razonable, de ensayos clínicos basados en los descubrimientos científicos.
Fuente bibliográfica
Treatment Possibilities for Retinitis Pigmentosa
Samuel G. Jacobson, M.D., Ph.D., and Artur V. Cideciyan, Ph.D.
From the Scheie Eye Institute, University of Pennsylvania, Philadelphia.
N Engl J Med 2010; 363:1669-167