Progresión tumoral, un problema de plasticidad
El melanoma maligno es un tumor agresivo de origen neuroectodérmico que se puede curar si se extirpa en una etapa temprana, sin embargo, una vez difundido a órganos distantes, la supervivencia media cae por debajo de 9 meses. La gran heterogeneidad intratumoral del cáncer unido a su potencial de auto-renovación, le permiten no seguir el modelo de células madre del cáncer, donde una sola "célula madre" maligna se reproduce para generar nuevas troncales y diferenciarse para ocasionar una nueva población tumoral. En vez esto, todas las células del melanoma tienen el potencial de ser células madre y ser capaces de inducir nuevos tumores. Estos hallazgos han revelado la particular biología del melanoma, y sugieren que esta afección requiere un nuevo enfoque terapéutico.
La visión tradicional del cáncer sostiene que éstos surgen a raíz de una acumulación al azar de acontecimientos malignos, por ejemplo, mutaciones, que gradualmente confieren suficientes ventajas para que una célula pueda crecer sin control. En la última década, los científicos han desarrollado el concepto de células madre del cáncer que explica cómo el lento crecimiento y la persistencia de las células madre permiten que los tumores persistan después del tratamiento. El melanoma, por su parte, parece seguir una tercera vía, la troncalidad dinámica, donde el comportamiento de las unidades progenitoras no se limita solamente a las células madre.
Progresión del cáncer
Ubicadas en un lugar oscuro de cada célula cancerígena están las mutaciones genéticas que les confieren el potencial maligno. Sin embargo, dentro de un tumor, esta capacidad no puede manifestarse en todas las células al mismo tiempo. Recientemente, Alexander Roesch y colegas (Cell 2010;141(4):583-594) han destacado la importancia de los factores no genéticos en la regulación del comportamiento maligno de las células cancerígenas, una observación que presenta desafíos y oportunidades para el tratamiento de los pacientes.
Mejor que nadie, los patólogos saben que en muchos tumores las células del cáncer no son todas iguales. La base de esta heterogeneidad celular ha intrigado a los biólogos debido a la posibilidad que las diferencias fenotípicas pueden ser indicativas de las disparidades funcionales en la capacidad de las células cancerígenas para propagar la enfermedad. Esta posibilidad tendría profundas implicaciones para el tratamiento porque las células cancerosas funcionalmente distintas posiblemente necesiten ser enfrentadas de otra manera.
La mejor explicación aceptada para dicha heterogeneidad es la evolución clonal. Esto ocurre cuando cada célula cancerosa adquiere mutaciones genéticas adicionales que alteran no sólo su fenotipo, sino también su potencial de malignidad, lo que resulta en la selección darwiniana de algunos clones durante la progresión de la enfermedad. Otra explicación es el modelo de células madre del cáncer, en que las unidades cancerígenas renuevan su propio potencial maligno, así como generan células cancerosas fenotípicamente distintas que pierden su efecto dañino a través de cambios epigenéticos estables. Una característica distintiva de estos dos modelos es la irreversibilidad de los cambios moleculares que dan lugar a diferencias fenotípicas y funcionales entre células.
En vista de las conclusiones del grupo de A. Roesch, habría que añadir otra forma de complejidad a la comprensión de tal heterogeneidad: la influencia de los factores reversibles. Estos investigadores identificaron sub-poblaciones en líneas celulares de melanoma que se distinguían por las diferentes cinéticas de proliferación y por la disparidad en la expresión de JARID1B, enzima que regula la expresión génica mediante la eliminación de grupos de metilo de un residuo de lisina en la histona 3. Aunque sólo una minoría de las células de melanoma examinadas expresaron JARID1B, la inhibición de la expresión JARID1B en líneas celulares deterioró la formación tumoral en los estudios con xenotrasplantes (figura 1A). JARID1B, por tanto, mantuvo la actividad en estas células cancerígenas. Este hallazgo no sólo es compatible con la gran cantidad de literatura que implica a la modificación de las histonas en la regulación de la tumorogénesis, sino que también sugiere estrategias para tratar el melanoma mediante la inhibición de la función JARID1B.
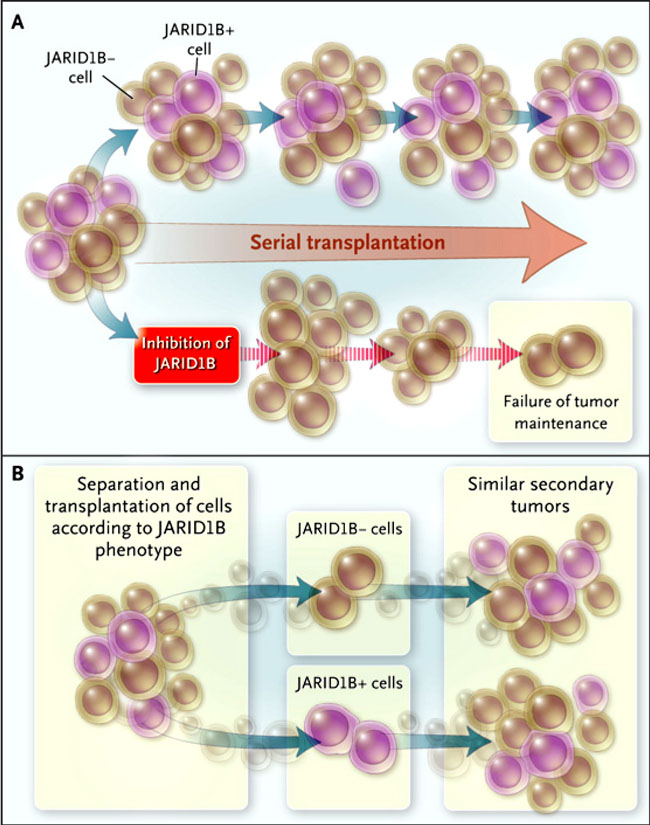
Aunque el potencial maligno subyacente de una célula cancerosa está determinado por mecanismos irreversibles, como la mutación genética, factores reversibles también pueden influir la capacidad para causar tumores. A. Roesch y colegas descubrieron recientemente que la enzima histona 3 desmetilasa, JARID1B, es fundamental para el mantenimiento de la actividad tumorigénica en líneas celulares de melanoma. El trasplante de células con JARID1B inhibió la pérdida de la capacidad del tumor (panel A). Sin embargo, se observó una heterogeneidad en la expresión de JARID1B en tumores secundarios derivados de JARID1B- o JARID1B+ (panel B), un hallazgo que indicó que la expresión de esta enzima no era estable, características hereditarias de estas células. Por lo tanto, mecanismos epigenéticos reversibles, pueden conducir a la progresión maligna.
La trama de esta historia se complicó considerablemente, sin embargo, cuando A. Roesch y colegas encontraron que la expresión JARID1B no era estable, purificaron células JARID1B- y JARID1B+, y cada una de la cuales produjo tumores que contenían células JARID1B- y JARID1B+ (fig. 1B). Por lo tanto, la expresión de JARID1B fue reversible, en contraste con los determinantes tumorigénicos que conducen la progresión del cáncer en la evolución clonal y en las células madre del cáncer. Aunque estos hallazgos necesitan ser confirmados en células no cultivadas de melanoma, los datos implican la plasticidad celular del cáncer como un determinante adicional de la heterogeneidad intratumoral.
Más importante aún, este trabajo indica que factores esenciales de la progresión del cáncer pueden ser blancos móviles que no están activos en todas las células en cada momento. Esta posibilidad tiene implicaciones fundamentales para el desarrollo de terapias dirigidas a mecanismos oncogénicos que no están activos simultáneamente. Centrarse exclusivamente en las células JARID1B+, podría no curar a los pacientes si estas unidades se reponen continuamente, derivadas de la persistencia de las células JARID1B-, después del tratamiento. Como mínimo, las terapias de este tipo tendrían que ser administradas a largo plazo, aumentando las preocupaciones acerca de su índice terapéutico y la posibilidad de desarrollar resistencia a los medicamentos.
¿Las células cancerosas podrían convertirse en blancos fáciles por obligarlas a convertirse en sensibles a la terapia? El uso de la histona deacetilasa (HDAC) podría representar un enfoque. Aunque los inhibidores de HDAC han demostrado actividad contra el cáncer, los estudios preclínicos sugieren que son potentemente sinérgicos cuando se combinan con terapias dirigidas a componentes específicos de la maquinaria oncogénica. Por ejemplo, en líneas celulares de cáncer, los inhibidores de HDAC han superado la resistencia a medicamentos mediada por otra histona desmetilasa de la familia JARID, JARID1A. Aún no se comprenden muy bien las modalidades de acción de los inhibidores de HDAC, pero es probable que incluyan la inducción de un estado epigenético en las células cancerosas que las hace vulnerables a los efectos de determinados agentes contra el cáncer.
Ya que las células del cáncer surgen y cambian como resultado de la mutación genética, se deben identificar y seleccionar los mecanismos que genéticamente confieren el determinado potencial maligno. Sin embargo, la investigación de A. Roesch pone de relieve otro medio por el cual las células cancerosas pueden cambiar durante la progresión de la enfermedad: la plasticidad epigenéticamente determinada. A medida que se clarifica la complejidad biológica de las células cancerosas, capa por capa, se hace evidente que los enfoques de tratamiento serán similarmente complejos. Otros estudios de plasticidad como causa de la heterogeneidad fenotípica y funcional de las células cancerosas tienden a convertir esta característica adaptativa de la enfermedad maligna en una oportunidad terapéutica.
Fuente bibliográfica
Moving Targets That Drive Cancer Progression
Mark Shackleton, M.D., Ph.D.
Melanoma Research Laboratory and Department of Hematology and Medical Oncology, Peter MacCallum Cancer Centre, East Melbourne, and the Department of Pathology, Faculty of Medicine, Dentistry and Health Sciences, University of Melbourne, Parkville.
N Engl J Med 2010; 363:885-886