ELA: triste y solitario final
La esclerosis lateral amiotrófica (ELA), a veces llamada enfermedad de Lou Gehrig, es la condición patológica de las neuronas motoras más frecuente en los adultos, y afecta aproximadamente a 1 de cada 100.000 personas. Sus síntomas incluyen la atrofia y parálisis de las extremidades inferiores y los músculos respiratorios a causa de la degeneración de las motoneuronas. En un 90 a 95 por ciento de todos los casos, la enfermedad ocurre aparentemente de forma aleatoria sin ningún factor de riesgo claramente asociado.
Actualmente no existe tratamiento eficaz. Así, la identificación de las vías de señalización celular y de los mediadores de la ELA sigue siendo un reto importante en la búsqueda de nuevas terapias.
Aminorar la esclerosis lateral amiotrófica
La esclerosis lateral amiotrófica (ELA) es una cruel enfermedad neurodegenerativa relacionada con la edad y por lo tanto es centro de intensa investigación. A veces es familiar, y cerca del 20% de estos casos son causados por mutaciones en SOD1, el gen que codifica la superóxido dismutasa 1. Recientemente se había creado un modelo de ratón para la enfermedad, que lleva una versión mutada del gen humano, sin embargo, ha sido cuestionado debido a que los tratamientos que mejoraban los síntomas en los ratones mutantes han fracasado en los seres humanos. Ahora, Andrew H. Williams y colaboradores (Science 2009; 326:1549-1554) sugieren un nuevo mecanismo de patogénesis, desplazando el centro de atención en el ratón mutante SOD1.
Esta condición humana es atribuida a la degeneración y pérdida de neuronas motoras, con el consiguiente mal funcionamiento muscular, causando debilidad, atrofia y, finalmente, la parálisis y muerte inevitable dentro de los 3 a 5 años después de la aparición sintomatológica. Williams y colegas, impresionados también por los informes que implicaban a los micro-ARN (miARN) en las respuestas al estrés en el músculo, observaron un incremento en la expresión de un micro-ARN específico (miR-206) para músculo en el ratón SOD1, y el aumento de la expresión de miR-206 coincidía con el inicio de los síntomas. Los miARN son pequeños ARN endógenos, alrededor de 22 nucleótidos de longitud, que se unen de manera específica al ARN mensajero e interfieren o nulifican su conversión a proteína. De esa manera, regulan la expresión génica y afectan a una amplia gama de procesos fisiológicos y de desarrollo.
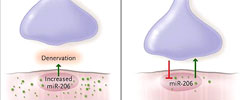
Williams y colegas reportaron que el corte del nervio ciático es seguido por un aumento de miR-206 en las fibras musculares, este incremento fue mayor en las fibras de contracción rápida que en las fibras de contracción lenta (figura 1). Los autores sugirieron que dos específicos factores musculares de transcripción, MyoD y miogenina, impulsaban la expresión de miR-206 mediante la unión al ADN de Mir206 (que codifica miR-206) y la activación de su transcripción. Estos hallazgos señalan que el flujo de la información molecular es bidireccional: fluye desde el músculo a la neurona, así como de la neurona al músculo.
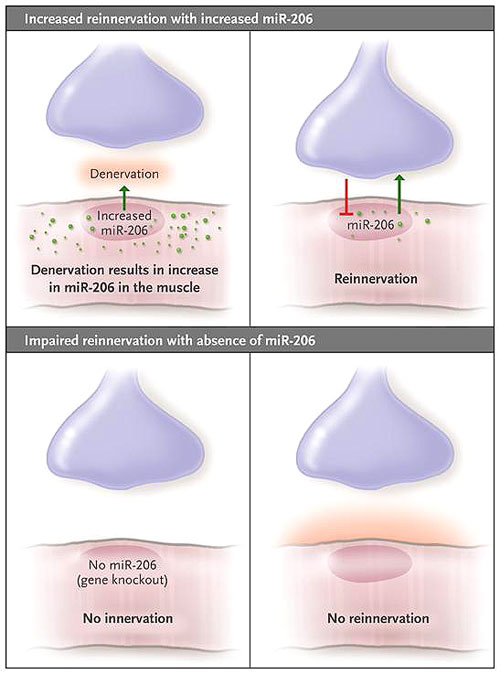
Un reciente estudio realizado por Andrew H. Williams y colaboradores implica un miARN (miR-206, que se expresa en el músculo esquelético) en el establecimiento y mantención integral de la unión neuromuscular. Un modelo de ratón para la esclerosis lateral amiotrófica que es deficiente en miR-206 (debido a la supresión de Mir206) demostró un retraso prolongado de la reinervación, en comparación con el modelo tipo salvaje Mir206.
Los síntomas de la enfermedad aparecieron en los ratones que habían sido modificados para carecer de miR-206 y llevar la proteína SOD1 mutante en la misma etapa de desarrollo que los ratones tipo salvaje con miR-206 y proteína SOD1 mutante. Sin embargo, estos ratones mostraron una progresión acelerada de la discapacidad y la disminución de la supervivencia como consecuencia de la acelerada atrofia muscular y parálisis.
Los autores no observaron ELA en animales deficientes en miR-206 y sin mutación SOD1. Además, no se contempló diferencia alguna en las uniones neuromusculares entre estos ratones y los tipo salvaje. Sin embargo, después de la denervación experimental, se vio comprometida la reinervación de las uniones neuromusculares en estos animales. Por otra parte, los sitios sinápticos se redujeron en número, las vesículas sinápticas no se agregaron normalmente, y los axones motores se extendieron más allá de las uniones sinápticas. Los autores concluyeron que miR-206 interviene en una señal derivada de las fibras musculares que es fundamental para la interacción con los axones regenerados. Propusieron que la reinervación compensatoria provocada por Mir-206 explicaba la falta de síntomas de la ELA temprana y que el número de músculos denervados finalmente aumenta de modo que la disminución de neuronas supervivientes no puede compensar el efecto.
El grupo de Andrew H. Williams pasó a delinear el camino a través del cual miR-206 pueda actuar en la mediación de la formación de la sinapsis neuromuscular. Para resumir, miR-206 indirectamente estimula la secreción de un factor de crecimiento de la proteína de unión a la matriz extracelular, que a su vez potencia los factores de crecimiento para promover la inervación presináptica en la unión neuromuscular.
Este estudio pone de manifiesto una vía que podría conducir a nuevos enfoques para tratar la ELA. Lo que se necesita ahora es la información acerca de la estabilidad post-mortem del miARN para que las mediciones puedan realizarse en seres humanos. Si se establece este equilibrio, se podría poner fin a la dependencia del ratón mutante SOD1.
Fuente bibliográfica
Ameliorating Amyotrophic Lateral Sclerosis
Lewis P. Rowland, M.D.
Neurological Institute, Columbia University Medical Center, New York, USA.
N Engl J Med. 2010 Mar 11; 362(10):953-4