SFRP, una declaración de independencia
Durante las últimas décadas, la vía de señalización Wnt se ha convertido en un actor clave en la embriogénesis y desarrollo postnatal, tanto en los procesos normales como patológicos. Las proteínas Wnt constituyen una gran familia de glicoproteínas secretadas que son capaces de activar un determinado número de cascadas de señalización celular en unión a las denominadas proteínas “Frizzled siete receptores transmembrana” y LRP5 o LRP6, que funcionan como co-receptores. Por su parte, la activación de la señal de Wnt es antagonizada por las “proteínas secretadas relacionadas a frizzled” (SFRP, por sus siglas en inglés), una familia de inhibidores de Wnt, cuyos miembros comparten la similitud de secuencia con el dominio rico en cisteína (DRC) que se encuentra en la región extracelular de Frizzled. Las SFRP se unen a los ligandos Wnt a través de su DRC, lo que impide su unión a los receptores Frizzled. En humanos, esta familia se compone de cinco miembros, SFRP1 a SFRP5, y ortólogos de estos genes se han encontrado en todas las especies de vertebrados.
Nuevos estudios demuestran que la SFRP2 de mamíferos puede actuar como un potenciador de la transformación de colágeno in vitro e in vivo, aumentando la fibrosis y el daño miocárdico. Estos hallazgos ponen de relieve la versatilidad biológica de los miembros de la familia de las SFRP.
Fibrosis e infarto de miocardio
Un reciente estudio de Koichi Kobayashi y colegas (Nat Cell Biol 2009; 11:46-55) demuestra que una proteína secretada por el organismo puede ser una interesante diana terapéutica para limitar la fibrosis y mejorar la función después de un infarto de miocardio. El cuadro clínico es un evento catastrófico, que provoca daños a la pared del corazón, por lo general, en la cámara del ventrículo izquierdo que bombea sangre a la circulación para perfundir otros órganos vitales. Esto puede conducir a insuficiencia cardíaca y, en consecuencia, a discapacidad o muerte. La herida causada por la necrosis y la apoptosis después de la oclusión de la arteria coronaria, desencadena un proceso de curación, resultando la cicatriz fibrótica y el restablecimiento de la función de bomba.
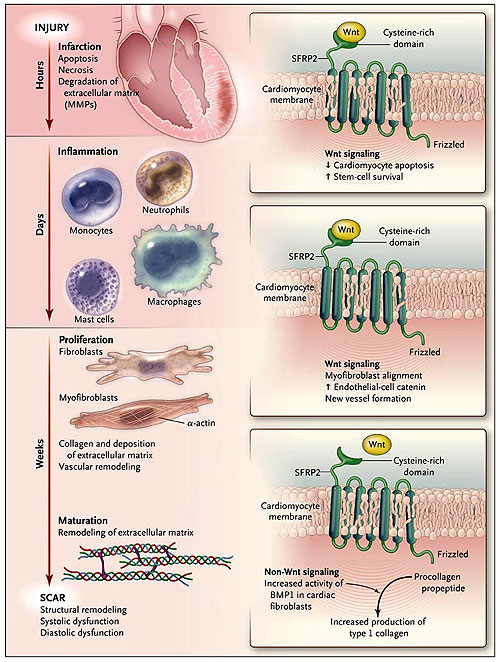
La mejora se logra a través de una serie de reacciones moleculares que involucran el tiempo de liberación de proteínas, tales como citoquinas, quimioquinas, metaloproteinasas de matriz, y los factores de crecimiento, el reclutamiento de células inflamatorias, la diferenciación de los macrófagos (a partir de monocitos), a mastocitos (precursores de células madre hematopoyéticas) y miofibroblastos (a partir de fibroblastos) y la formación de nuevos vasos y tejido cicatricial (figura 1). La matriz de metaloproteinasas degrada la matriz extracelular, lo que lleva al desvío de los cardiomiocitos y la ampliación cardíaca.

El daño miocárdico provoca una dinámica molecular y cambios celulares que generan cicatrices en el ventrículo, remodelación, fibrosis y disfunción. Durante el infarto agudo, mueren muchos cardiomiocitos, y aumentan los niveles de metaloproteinasas de la matriz (MMPs) afectando la matriz extracelular. Al mismo tiempo, la SFRP2 reprime la señalización de Wnt y, por tanto, la apoptosis de los cardiomiocitos, mejorando la supervivencia de las células madre. Durante la fase de proliferación y curación, SFRP2 continúa inhibiendo la señalización de Wnt, mediando el alineamiento de los miofibroblastos y la neovascularización. Al mismo tiempo y durante la fase de maduración, SFRP2 aumenta la producción de colágeno por los fibroblastos cardiacos a través de una vía independiente de Wnt por el incremento de la actividad de la proteína morfogenética ósea 1 (BMP1), dando lugar a una elevada producción de colágeno de tipo I.
Normalmente, la cantidad de colágeno y de fibrosis varía según la zona del infarto al final de la fase de curación. La fuerte remodelación de las paredes del músculo cardíaco involucra tanto al tejido dañado como al sano y se produce con el objetivo de preservar la función y, por tanto, la supervivencia. Sin embargo, cuando la zona afectada es grande, la curación y los mecanismos de reparación pueden ser agobiantes. También pueden entrar en un proceso de desregulación generando una remodelación cardíaca progresiva, fibrosis inadecuada, y aún más, la disfunción total. Por ejemplo, el exceso de fibrosis en la zona no infartada así como en la del infarto conlleva un aumento de la disfunción diastólica, la cual, a su vez, conduce a una mayor disfunción sistólica. Otra ruta que puede aparecer en la disfunción sistólica es la fibrosis defectuosa en la zona del infarto. La fibrosis defectuosa es una consecuencia de un depósito de colágeno débil o de una relativa escasez de colágeno maduro tipo I (que se opone a la distensión) y una abundancia de colágeno inmaduro tipo III (que es más distensible). La terapia antifibrótica durante el manejo del infarto puede tener un elevado costo.
K. Kobayashi colegas evaluaron el efecto del silenciamiento de la proteína secretada relacionada a frizzled (SFRP) en la fibrosis. Las SFRPs muestran un alto grado de conservación molecular en las especies y tienen un dominio rico en cisteína que une a los receptores transmembrana Wnt y, por tanto, inhiben la señalización intracelular de Wnt (la señalización Wnt modula los procesos de desarrollo, en parte por el control de la apoptosis). Varios estudios han sugerido que reprimir la señalización de Wnt por SFRP2 puede ser beneficiosa después del infarto de miocardio. Por ejemplo, durante el infarto agudo en ratas, se demostró que SFRP2 induce una mejor protección mediante la inyección de células madre y la inhibición de la apoptosis mediada por cardiomiocitos a través de la represión de la señalización de Wnt. Además, después del infarto, se encontró que SFRP2 regula a los miofibroblastos y modifica la neovascularización del infarto por la coacción de la señalización de Wnt (figura 1).
Sin embargo, las SFRPs también funcionan de manera independiente del mecanismo de señalización de Wnt. Estudios en peces cebra y en ranas han demostrado que SFRPs son capaces de modular la remodelación de los tejidos por aumento de la actividad de las metaloproteinasas, las cuales convierten el pro-colágeno en colágeno y, por tanto, modifican la matriz extracelular. El grupo de Kobayashi encontró que las SFRP2 de mamíferos aumentan las concentraciones de metaloproteinasas. Esto activa y eleva la trasformación de pro-colágeno en colágeno (figura 1). Los autores demostraron que en el tejido cardíaco recuperado (anteriormente lesionado a través de un infarto de miocardio) de ratones Sfrp2-, la deposición del colágeno y la fibrosis se redujo, de paso mejorando la función sistólica. También observaron una correlación espacial y temporal entre la expresión de la proteína morfogenética ósea 1 (una metaloproteinasa tipo TLD) y de las SFRP2 durante y después de la mejora del infarto en ratones tipo salvaje. Sin embargo, ellos no midieron los niveles de colágeno total y los diferentes tipos de colágeno en las zonas infartadas y no infartadas, ni tampoco realizaron estudios más allá de la fase de curación.
Aunque los antagonistas de SFRP2 teóricamente podrían utilizarse para controlar y mejorar la función de la fibrosis en el corazón infartado, como sugieren los autores, se requieren estudios adicionales antes de que tal enfoque sea considerado en el ámbito clínico. En general, es prudente actuar con cautela cuando se utilizan agentes antifibróticos durante el manejo de los efectos adversos del infarto, y evaluar la seguridad a largo plazo.
Fuente bibliográfica
Limiting Fibrosis after Myocardial Infarction
Bodh I. Jugdutt, M.D., D.M.
Division of Cardiology, Department of Medicine and Cardiovascular Research Group, Faculty of Medicine, University of Alberta, Edmonton, Canada.
N Engl J Med. 2009 Apr 9;360(15):1567-9