Cicatrización cardíaca, ¿proceso reversible?
Un reciente informe dirigido por investigadores de Beth Israel Deaconess Medical Center (BIDMC) ayuda a explicar el origen de la fibrosis cardíaca, un endurecimiento del músculo del corazón que conduce a una variedad de enfermedades, en particular a la insuficiencia cardíaca. En ensayos realizados en animales, se ha demostrado que la molécula morfogénica ósea conocida como rhBMP7, puede invertir el proceso de la fibrosis cardíaca, ofreciendo la posibilidad de un objetivo terapéutico para esta condición debilitadora.
Las patologías cardíacas son la principal causa de muerte en el mundo occidental, explica Elisabeth Zeisberg, una destacada doctora en la Division of Matrix Biology en BIDMC e instructora en la Escuela de Medicina de la Universidad de Harvard. Para ella, la mayoría de las personas que sufren de enfermedades al corazón han desarrollado la cicatrización del tejido cardíaco, conocida como fibrosis. La fibrosis se desarrolla cuando el proceso natural de la curación de una herida se altera erróneamente. En condiciones normales, células especializadas conocidas como fibroblastos depositan capas de proteínas de colágeno para formar una cicatriz y, en consecuencia, permitir sanar las heridas. Sin embargo, en circunstancias anormales, y por razones que se desconocen, la excesiva producción proteica de la matriz, como el colágeno, resulta en la cicatrización patológica, o fibrosis. En el corazón, la acumulación de matriz deja el órgano rígido e inflexible, incapaz de funcionar correctamente y relajarse.
El grupo de la Dra. Zeisberg ha especulado que una forma especializada de transición epitelio-mesenquimal, conocido como transición endotelio mesénquima, podría ser el mecanismo detrás de este giro de acontecimientos. También ellos han observado que las proteínas rhBMP-7 tienen una tremenda capacidad para recuperar la función del corazón dañado. Estos resultados proporcionan pruebas convincentes de que el proceso de la fibrosis puede ser invertido y ofrece la posibilidad de nuevas terapias para pacientes que han desarrollado la fibrosis cardíaca como consecuencia de un infarto de miocardio, hipertensión, enfermedades valvulares o por trasplante de corazón.
Cicatrización cardíaca y transición epitelio-mesenquimal
Durante décadas se ha creído que la fibrosis cardíaca, o cicatrización cardíaca, es un proceso irreversible que conduce a alteraciones funcionales del miocardio y, en definitiva, a anomalías de la contractilidad cardíaca (disfunción sistólica) y de relajación (disfunción diastólica). Un estudio reciente realizado por Elisabeth M. Zeisberg y colegas indica que, al igual que muchas "cosas seguras" en medicina y en ciencia, nos obliga a repensar si el desarrollo de la fibrosis y sus efectos adversos que tienen sobre la función cardíaca son irreversibles. La fibrosis cardíaca tiene varias causas, entre ellas la isquemia y el infarto, las miocardiopatías y miocarditis, todas ellas con seguimiento a corto y largo plazo debido a la disfunción de la bomba y a la rigidez miocárdica. Los tratamientos se prescriben de acuerdo a los síntomas, sin embargo, un alto porcentaje de las personas afectadas sufren insuficiencia cardíaca o muerte súbita de causa cardíaca.
La fibrosis se asocia con alteraciones de la estructura normal del corazón a nivel de los cardiomiocitos y a una deposición y acumulación excesiva de proteínas de la matriz extracelular. Los mediadores celulares de la fibrosis predominantemente son los fibroblastos, cuyo origen es incierto. En general, se supone que los fibroblastos adultos se originan de las células embrionarias mesenquimatosas y que aumentan en número únicamente como consecuencia de la proliferación de los fibroblastos adultos residentes. Esta hipótesis ha sido cuestionada por un estudio que sugiere que, durante los procesos fibrosantes, los fibroblastos derivados de la médula ósea y las células epiteliales también contribuyen a la acumulación de fibroblastos, por medio de un proceso llamado transición epitelio-mesenquimal. Esta transición es fundamental para el desarrollo embrionario del corazón, ya que es el medio por el cual las células mesenquimales que forman el “cojín” auriculoventricular, que son las formas primordiales de las válvulas y los septos del corazón adulto, derivan del endocardio. Las señales inductivas, como el factor de crecimiento transformador beta (TGF-beta) y las proteínas morfogénicas óseas, están diseñadas para regular la transición.
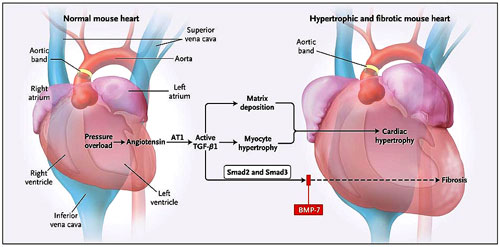
El grupo de Zeisberg evaluó la hipótesis de que la fibrosis cardíaca se produce, en parte, por medio de la acumulación de fibroblastos aportados por la transición epitelio-mesenquimal. Al colocar bandas aórticas en el corazón de ratones y luego marcar y rastrear permanentemente las células de origen endotelial, los investigadores demostraron que éstas se sometían a la transición epitelio-mesenquimática durante la fibrosis cardíaca contribuyendo a la reserva total de fibroblastos cardíacos. Además, demostraron que los niveles de expresión de los marcadores mesenquimales eran mucho mayores, y que los marcadores de las células endoteliales eran sustancialmente inferiores en el corazón fibrótico que en el no fibrótico. Los autores observaron que TGF-beta1, un conocido promotor de la fibrosis cardíaca, es capaz de inducir la transición epitelio-mesenquimal, mientras que la proteína morfogenética ósea 7, un antagonista de la vía de TGF-beta, conserva el fenotipo endotelial y revierte o impide la transición inducida por TGF-beta1 (y, por lo tanto, la fibrosis, figura 1). Además, la investigación puso de manifiesto que es posible reducir la fibrosis cardíaca inducida por TGF-beta1 en ratones con sólo la mitad del nivel normal del factor de transcripción Smad3, un miembro del proceso de señalización de TGF-beta que es activado por TGF-beta1.

Elisabeth Zeisberg y colaboradores demostraron recientemente que la transformación del factor de crecimiento beta1 (TGF-beta1) impulsa la transición endotelio-mesenquimal. También manifestaron que esta transición impulsa la fibrosis cardíaca, inducida por bandas aórticas, con la consiguiente hipertrofia. TGF-beta1 afecta a esta transición mediante la activación de las moléculas Smad2 y Smad3; miembros de la familia TGF, la proteína morfogenética ósea 7 (BMP-7), controla esta transición y la protege contra la fibrosis inducida cuando se administra antes del bandeo. AT1 denota la angiotensina II tipo 1.
La superfamilia TGF-beta tiene más de 40 miembros, incluido TGF-beta, factor de crecimiento transformador tipo B, y proteínas morfogénicas óseas. Los miembros de esta superfamilia parecen conducir a la fibrosis en el corazón, los riñones, los pulmones y el hígado. Se han descrito tres diferentes isoformas de TGF beta, con TGF-beta1 encontrándose predominantemente en el sistema cardiovascular. TGF-beta1 se sintetiza como una proteína inactiva anclada en la matriz extracelular, y es estimulada por la angiotensina II, que la convierte en la forma biológicamente activa (figura 1). La forma activa se une a los receptores y estimula a Smad2 y Smad3 (que también participan en las vías de señalización de las proteínas morfogénicas del hueso). La identificación de la sobre regulación de TGF-beta1, Smad2 y Smad3 en los corazones fibróticos bandeados apoya la idea de que esta vía es importante para el desarrollo y la progresión de la cicatrización cardíaca y la hipertrofia del órgano (figura 1).
Para cumplir con los postulados de Koch, los autores evaluaron si la obstrucción de la vía previene o invierte el fenotipo clínico, y demostraron elegantemente que la proteína morfogenética ósea 7, un miembro de la superfamilia TGF-beta, bloquea de hecho la fibrosis. En estudios que han involucrado a células endoteliales coronarias de humanos adultos incubadas con TGF-beta1, un fenotipo de fibroblastos, la exposición a la proteína morfogenética ósea 7 inhibió fuertemente la transición epitelio-mesenquimal inducida por TGF-beta1. Los autores también administraron la proteína ósea un día antes de aplicar las bandas aórticas y posteriormente cada día durante los próximos 28 días; observando la reducción de de la fibrosis en un 40%, en comparación con el descenso visto en los ratones bajo placebo. Por otra parte, la hipertrofia también fue inhibida en los ratones tratados.
La labor de los colegas de Elisabeth Zeisberg proporciona una base molecular para el desarrollo de la fibrosis cardiaca, demostrando una relación entre la fibrosis y la hipertrofia, e identificando una molécula que podría prevenir o revertir la cicatrización cardíaca. Nuevos tratamientos, quizás en combinación con el uso de antiguos medicamentos que intervienen en la vía de TGF-beta, como los inhibidores de la enzima convertidora de la angiotensina, podrían ayudar en la lucha para preservar la función cardíaca en pacientes con cardiopatía isquémica, miocarditis o miocardiopatías congénitas.
Fuente bibliográfica
Scarring in the Heart — A Reversible Phenomenon?
Jeffrey A. Towbin, M.D.
Section of Pediatric Cardiology, Department of Pediatrics, Baylor College of Medicine and Texas Children's Hospital, Houston, USA.
N Engl J Med. 2007 Oct 25;357(17):1767-8