Genética y enfermedades, más diferentes de lo que creíamos
INTRODUCCIÓN
Las nuevas y recientes investigaciones están terminando con la clásica idea de que los códigos genéticos de dos personas son idénticos en un 99.9%. Además, también cuestionan la visión de que cada persona hereda de sus progenitores un juego completo de genes, con una sola copia de cada uno, en el momento de la fecundación. Los últimos reportes al respecto, demuestran que cerca de unos 2.900 genes (de los 20.000 a 25 mil que hay en todo el genoma) son susceptibles de estar repetidos, en algunos casos hasta 14 veces, o simplemente borrados. También se minimiza el planteamiento de que las diferencias genéticas entre personas se deben principalmente a cambios que afectan a un solo nucleótido, como por ejemplo, en el caso de los denominados polimorfismos de nucleótido único (SNP, según su sigla en inglés). Estos cambios han sustentado la búsqueda de las causas genéticas para las enfermedades desde el término del Proyecto Genoma Humano (PGH), en el año 2000.
Los nuevos análisis revelan que lo que hace diferente a cada persona se debe, en gran parte, a zonas del genoma que afectan a miles de nucleótidos. Estos bloques funcionan como láminas en el ADN: se pueden tener repetidos, incluso varias veces, invertidos o pueden faltar. Por consiguiente, los individuos suelen tenerlos en números distintos. De ahí su nombre variantes de número de copia (o CNV, según su sigla en inglés). El resultado más importante de estos descubrimientos, es que estas variantes no son una rareza, sino que abarcan el 12% del genoma humano. En este porcentaje se encuentran 285 de los 1.961 genes relacionados con enfermedades que se han identificado hasta la fecha. Entre ellos destacan genes que tienen que ver con enfermedades frecuentes como la arteriosclerosis, el Alzheimer, el Parkinson, la esquizofrenia o las cataratas. Por ejemplo, el número de enfermedades monogénicas es mayor de seis mil. Aunque cada una de ellas suele ser poco frecuente pero, en su conjunto, constituyen un problema médico de primera magnitud debido a que muchas causan graves trastornos a los pacientes y representan una gran carga física y emocional, cuando no socioeconómica, para las familias.
Se sabe que, cuando un gen está repetido, se eleva la cantidad de proteína que genera afectando de este modo la posibilidad de padecer ciertas enfermedades. En algunos casos, tener más proteína aumenta el riesgo de una enfermedad y en otros casos lo reduce. Muchos de los genes mutantes responsables de patologías neurológicas se han localizado en el genoma humano y se han caracterizado e identificado los productos génicos. Hoy en día es posible determinar la enfermedad preclínica, realizar la identificación de individuos portadores y ofrecer un diagnóstico prenatal y, esperanzadoramente, obtener tratamientos eficaces en un plazo de tiempo relativamente corto.
En definitiva, esta “variación estructural” del ADN afecta la regulación de muchos genes, el Dr. James Lupski explora a continuación las implicaciones clínicas de estos grandes cambios.
Variación estructural del genoma humano
El término del proyecto del genoma humano fue una hazaña notable que arrojó información privilegiada para los estudios comparativos de unos 3 mil millones de bases. Una analogía frecuentemente utilizada para conceptuar la información genética humana es la de una enciclopedia, en la cual cada volumen del sistema representaría uno de los 23 pares de cromosomas humanos. Las secciones dentro de cada volumen representarían (aproximadamente) los 25.000 genes del genoma de la especie humana, y las letras del alfabeto serían las bases individuales del ADN que codifican aminoácidos específicos formadores de la estructura de las proteínas. Hasta la fecha, el modelo de la medicina molecular que se enseña en cada facultad se basa en la enfermedad celular, en la cual el tipo de mutación predominante es un cambio en el par de bases (según la analogía de la enciclopedia, sería la substitución de una sola letra), que altera la secuencia y los resultados de codificación en la síntesis de una proteína mutante. En este modelo, la variación genética entre individuos y entre poblaciones se presenta por la variación de las bases, también conocida como polimorfismos de un solo nucleótido.
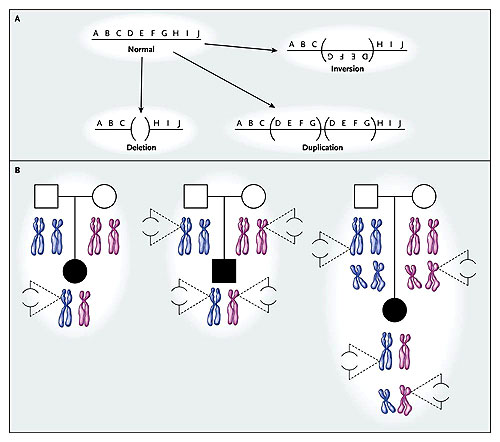
Sin embargo, parece evidente que el resultado final de la secuenciación del ADN humano y el desarrollo de nuevas tecnologías para detectar el grado y posición de las alteraciones genómicas dentro de un solo genoma ha detectado que se han suprimido o duplicado grandes fragmentos de nuestro ADN. Este re-arreglo genómico puede cambiar el número de copias de los genes alterando las zonas afectadas y por consiguiente la regulación génica. Un reciente estudio de Redon y colegas (Nature. 2006 Nov 23; 444(7118):444-54) proporciona el primer mapa de variación genética respecto al número de copias en todo el genoma humano. El mapa muestra que esta variación puede abarcar varios nucleótidos (por analogía, las letras) al igual que cambios individuales representando así una fuente importante de variación. De hecho, existe un enorme grado de variación estructural en el genoma humano entre personas relacionadas y entre poblaciones, causada por grandes deleciones y duplicaciones de los segmentos genómicos (figura 1). Redon y colaboradores identificaron variaciones en el número copias en 1.400 regiones que se traslapaban con el 14.5% de los genes implicados en enfermedades humanas, enumeradas en la base de datos OMIM (Online Mendelian Inheritance in Man, www.ncbi.nlm.nih.gov/omim).
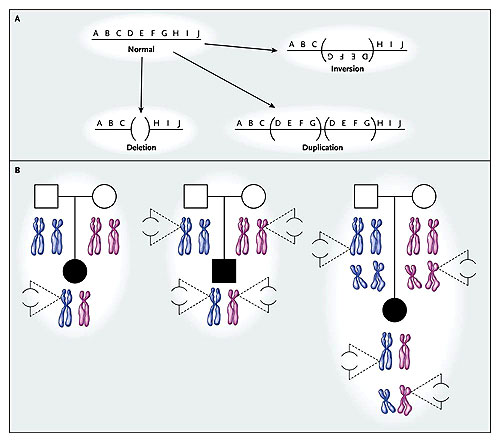
El panel A señala la variación estructural del genoma humano. Las líneas horizontales con letras sobre ellas representan el genoma. Se grafica un solo cromosoma. Las deleciones y duplicaciones dan lugar a variaciones en el número de copias de las secuencias que contienen los cambios. El panel B muestra tres mecanismos potenciales por los cuales las variaciones en el número de copias o las combinaciones de tales variaciones dan lugar a un fenotipo esporádico. Los cuadrados representan a varones, los círculos a hembras, y los símbolos oscuros a personas con el fenotipo de la enfermedad. Debajo de los pedigríes, se observan los cromosomas paternos (azules) y maternos (rosados). Las líneas discontinuas señalan la ampliación de regiones sub-microscópicas, las líneas verticales el genoma, y en paréntesis una zona suprimida del genoma que causa una variación en el número de copias. En el primer caso (izquierda), una nueva deleción dominante ocurre en el cromosoma heredado del padre; las nuevas mutaciones pueden causar rasgos esporádicos con más frecuencia que aquellas mutaciones en un solo punto. En el segundo caso (centro), una deleción en el mismo lugar se hereda recesivamente de cada padre. En el tercer caso (derecha), se muestra la herencia de una deleción en diversos lugares y en dos cromosomas diferentes.
Probablemente, muchas alteraciones en el número copias son benignas, pero otras variaciones específicas se asocian a condiciones mendelianas comunes (en un solo lugar), tales como la acromatopsia, la enfermedad Charcot-Marie-Tooth de tipo 1A y a otros desórdenes del sistema nervioso. La variación en las copias también puede influenciar la susceptibilidad a complejas enfermedades como la glomerulonefritis asociada con lupus y a la infección con el virus de la inmunodeficiencia humana, así como a la enfermedad de Parkinson, Alzheimer y enfermedad de Crohn. La aplicación clínica de matrices genómicas, “laminillas” de ADN que detectan cambios en el número de copias de partes específicas del genoma, ha permitido detectar muchas deleciones y duplicaciones sub-microscópicas que causan complejos síndromes de retraso mental. La demostración de la existencia de aberraciones en una área determinada del gen (número de copias presentes en una célula o en un núcleo) como un mecanismo de enfermedad abre nuevos caminos para desarrollar de forma más fácil tratamientos en los cuales el objetivo no sería corregir las proteínas disfuncionales o mutantes sino que por el contrario modificar su anormal estructura génica.
Encontrar un alto grado de variación en el número de copias en el genoma humano también cambiará nuestras investigaciones sobre las causas de las enfermedades genéticas. De ahora en adelante, los estudios genéticos deben incorporar una evaluación de las variaciones de la cantidad de copias en la población de estudio para determinar si una alteración individual del número de copias, más que un polimorfismo de un solo nucleótido, pudiera ser responsable del rasgo que se investiga. Además, una enfermedad esporádica (es decir que se da por casos aislados dentro de una familia) puede resultar de una nueva alteración genómica que causa la variación en el número de copias o de una combinación de dos o más variaciones heredadas a partir de ambos padres, en lo cual la variación sin combinar no proporciona una carga genética lo suficientemente grande para causar la enfermedad (figura 1). Incluso, uno podría especular que la variación en el número de copias es la base de rasgos normales y comunes tales como los implicados en el comportamiento.
Fuente bibliográfica
Structural variation in the human genome
James R. Lupski, M.D., Ph.D.
Department of Molecular and Human Genetics, Baylor College of Medicine and Texas Children's Hospital, Houston, USA.
N Engl J Med. 2007 Mar 15; 356(11):1169-71